

Semiempirical Quantum Mechanical Methods for Noncovalent Interactions for Chemical and Biochemical Applications
- PMID: 27074247
- PMCID: PMC4867870
- DOI: 10.1021/acs.chemrev.5b00584
Semiempirical Quantum Mechanical Methods for Noncovalent Interactions for Chemical and Biochemical Applications
Abstract
Semiempirical (SE) methods can be derived from either Hartree-Fock or density functional theory by applying systematic approximations, leading to efficient computational schemes that are several orders of magnitude faster than ab initio calculations. Such numerical efficiency, in combination with modern computational facilities and linear scaling algorithms, allows application of SE methods to very large molecular systems with extensive conformational sampling. To reliably model the structure, dynamics, and reactivity of biological and other soft matter systems, however, good accuracy for the description of noncovalent interactions is required. In this review, we analyze popular SE approaches in terms of their ability to model noncovalent interactions, especially in the context of describing biomolecules, water solution, and organic materials. We discuss the most significant errors and proposed correction schemes, and we review their performance using standard test sets of molecular systems for quantum chemical methods and several recent applications. The general goal is to highlight both the value and limitations of SE methods and stimulate further developments that allow them to effectively complement ab initio methods in the analysis of complex molecular systems.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
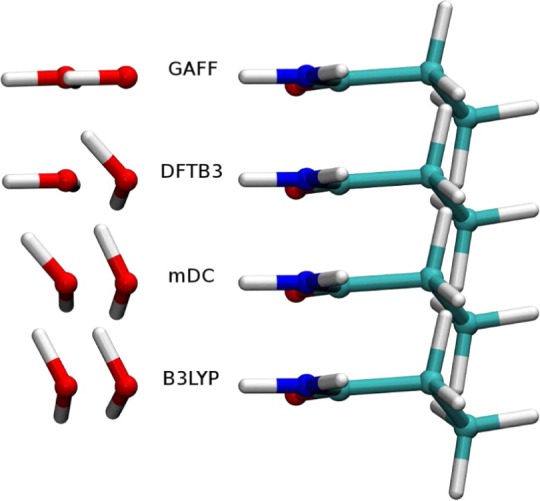
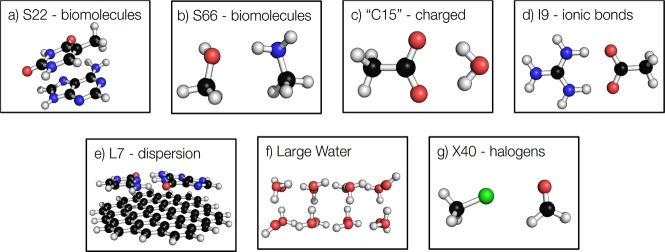
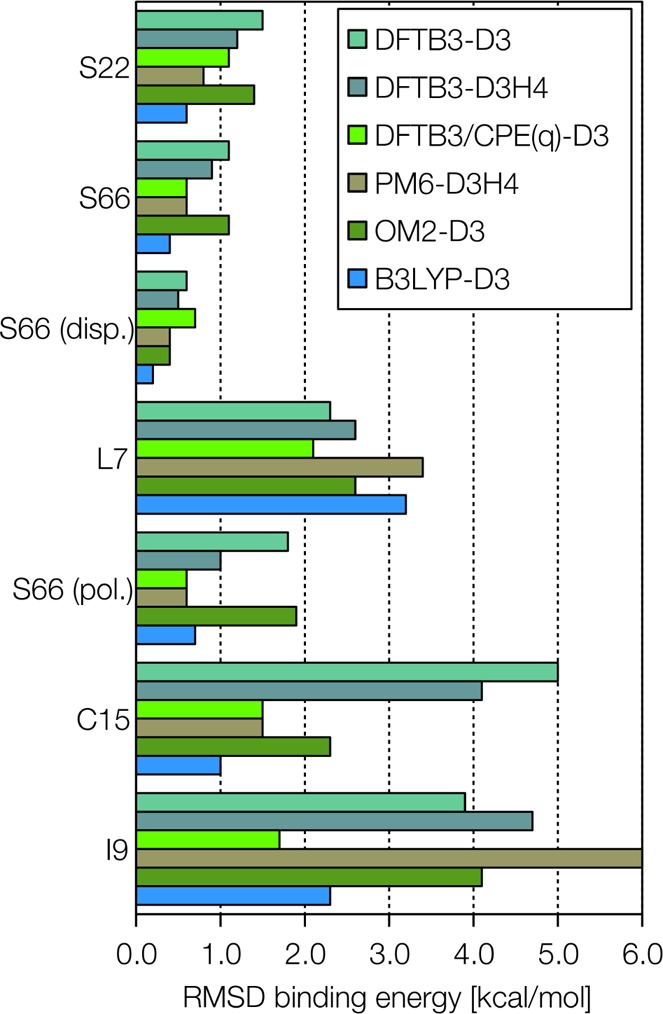

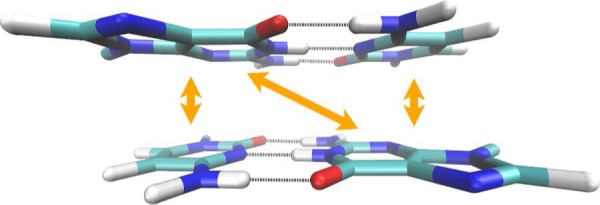
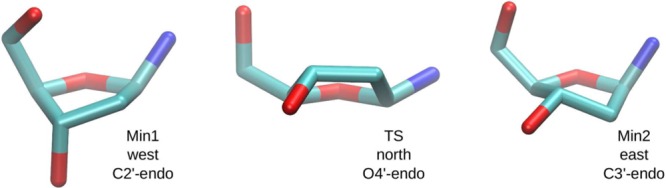
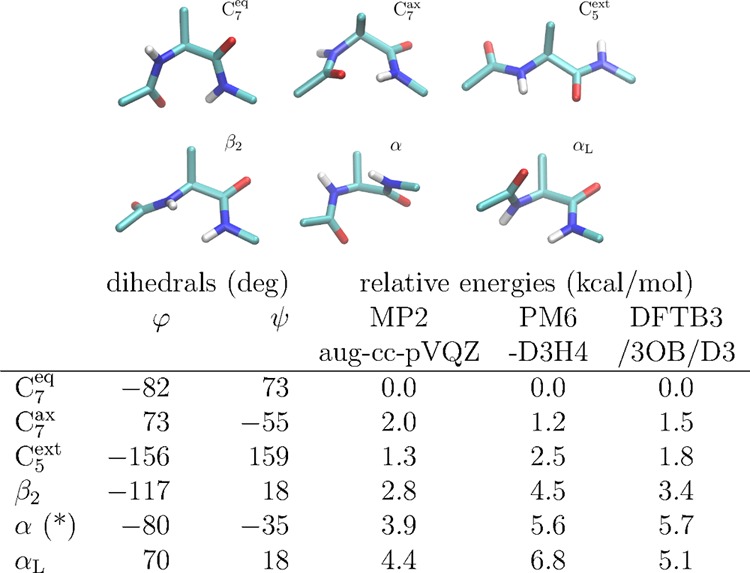
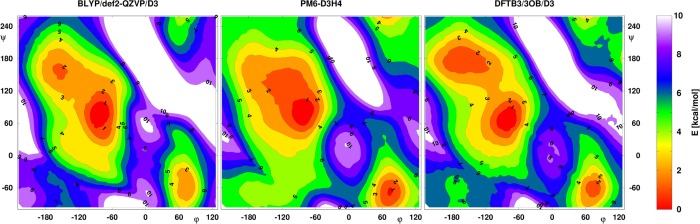
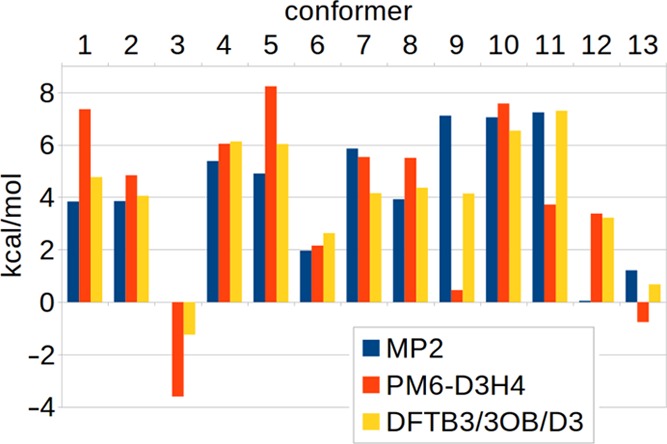
References
-
- Banks J. L.; Kaminski G. A.; Zhou R. H.; Mainz D. T.; Berne B. J.; Friesner R. A. Parametrizing a Polarizable Force Field From Ab Initio Data. I. The Fluctuating Point Charge Model. J. Chem. Phys. 1999, 110, 741–754. 10.1063/1.478043. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
