

NFκB is a central regulator of protein quality control in response to protein aggregation stresses via autophagy modulation
- PMID: 27075172
- PMCID: PMC4884063
- DOI: 10.1091/mbc.E15-12-0835
NFκB is a central regulator of protein quality control in response to protein aggregation stresses via autophagy modulation
Abstract
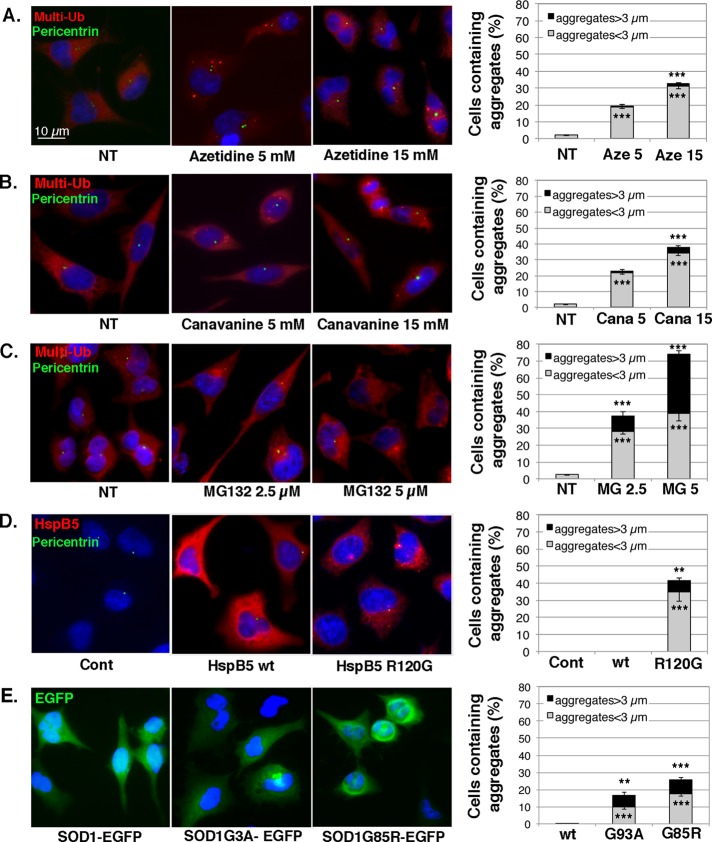
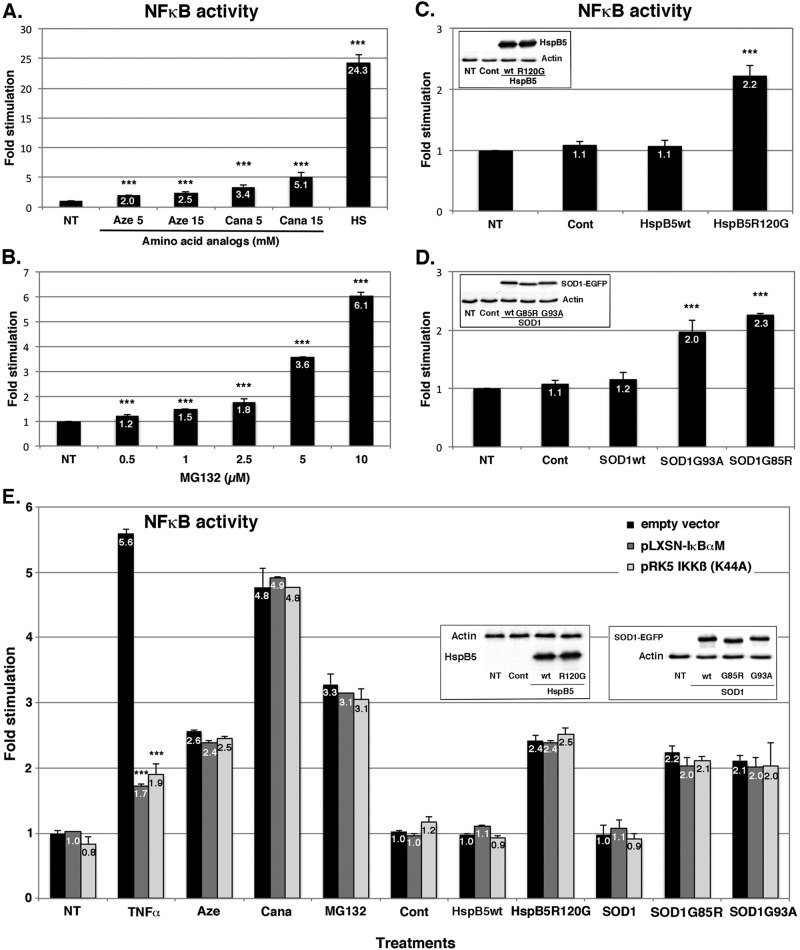
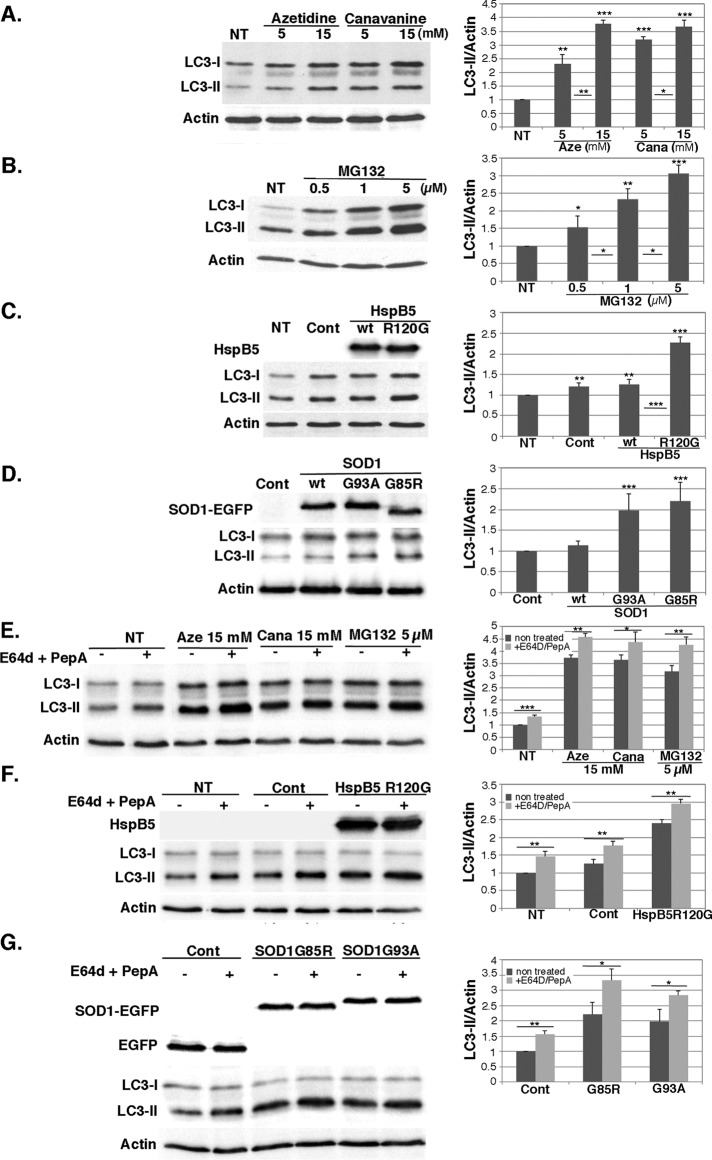
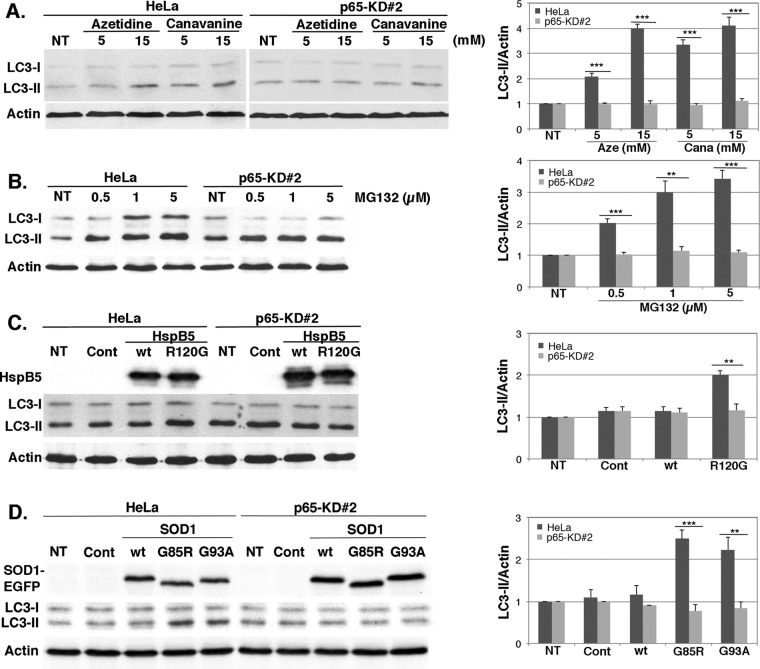
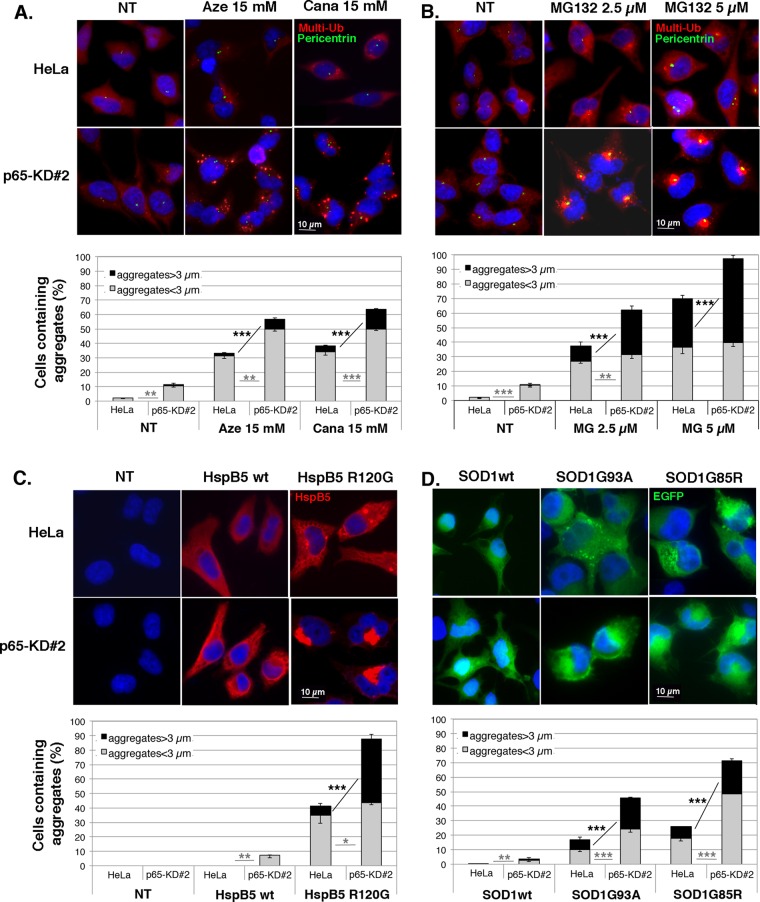
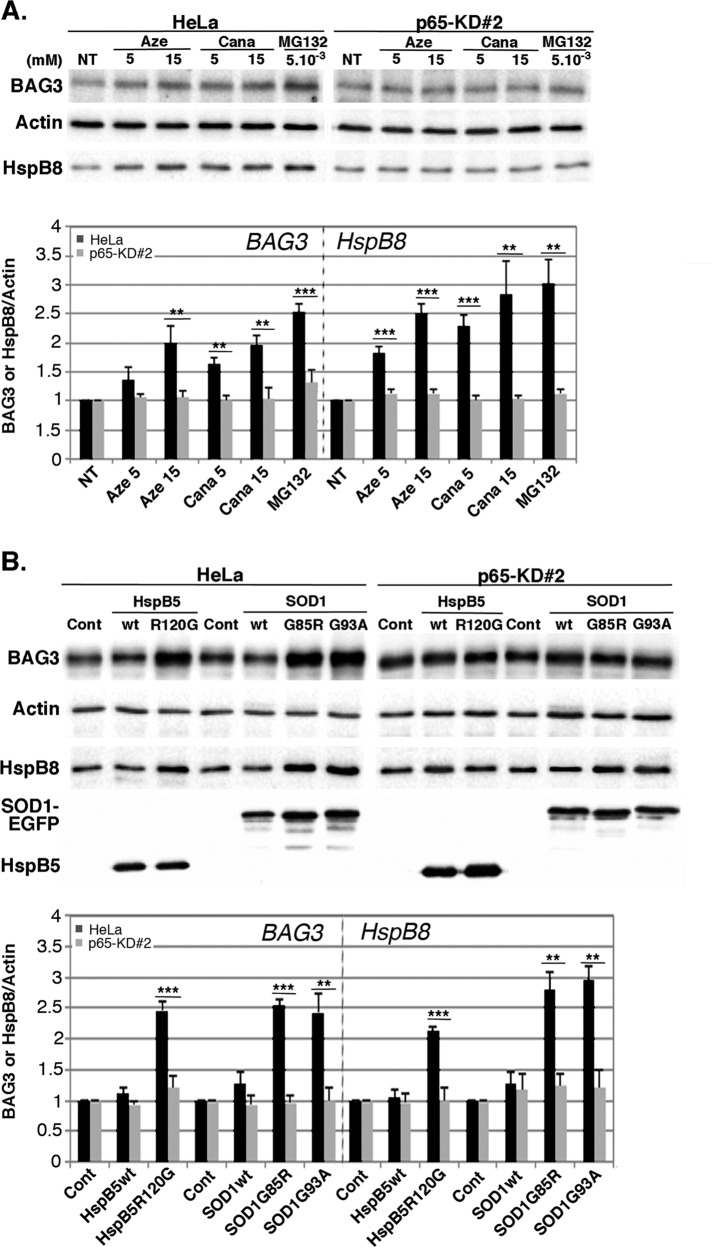
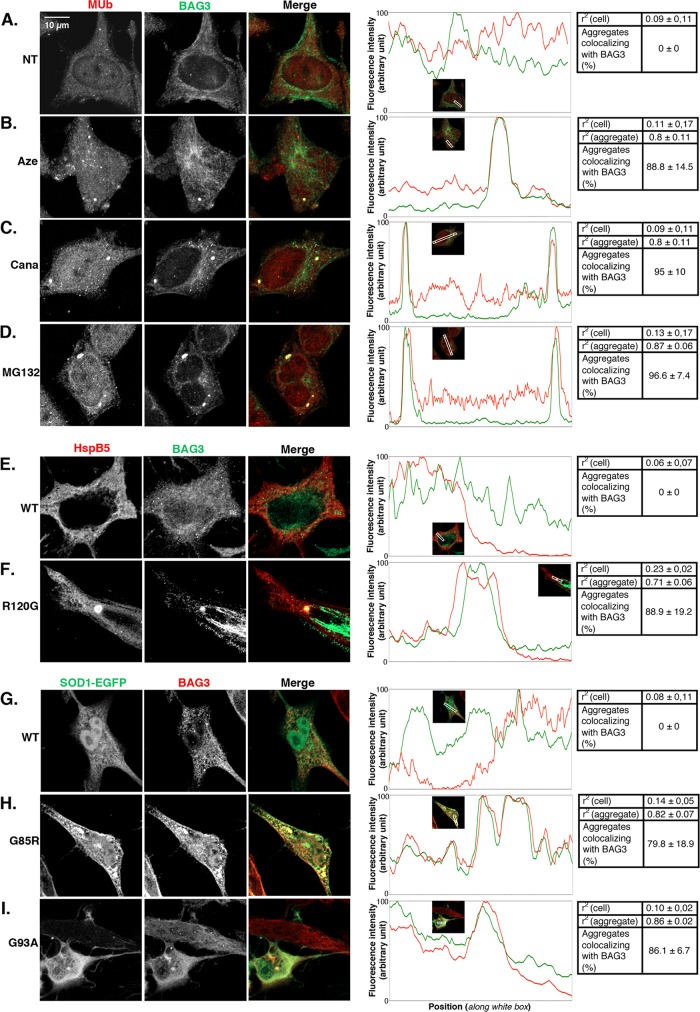
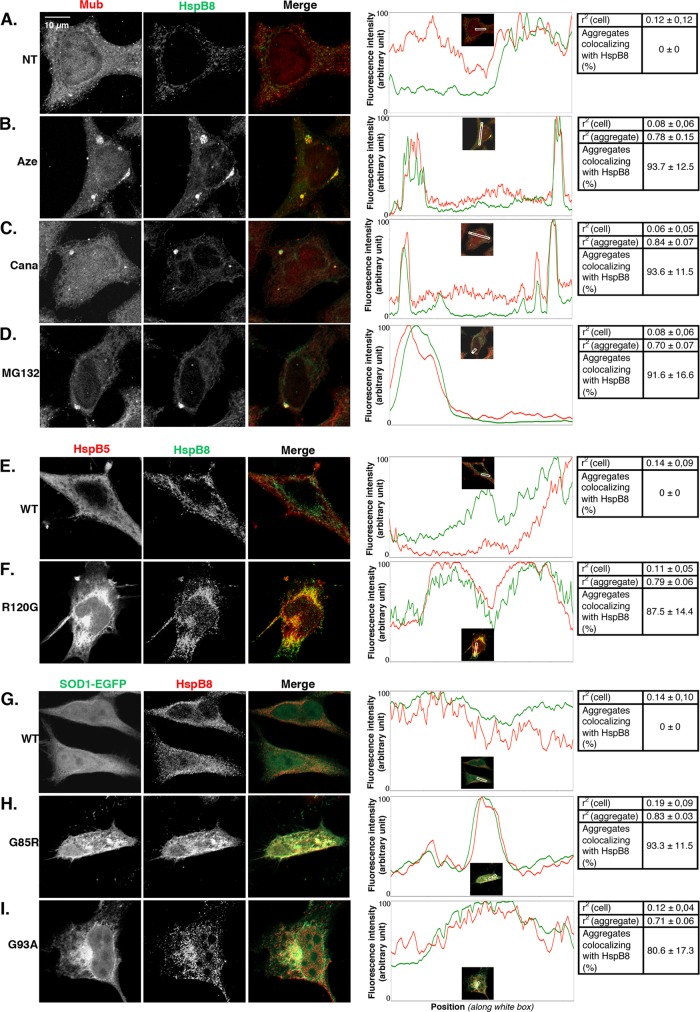
During cell life, proteins often misfold, depending on particular mutations or environmental changes, which may lead to protein aggregates that are toxic for the cell. Such protein aggregates are the root cause of numerous diseases called "protein conformational diseases," such as myofibrillar myopathy and familial amyotrophic lateral sclerosis. To fight against aggregates, cells are equipped with protein quality control mechanisms. Here we report that NFκB transcription factor is activated by misincorporation of amino acid analogues into proteins, inhibition of proteasomal activity, expression of the R120G mutated form of HspB5 (associated with myofibrillar myopathy), or expression of the G985R and G93A mutated forms of superoxide dismutase 1 (linked to familial amyotrophic lateral sclerosis). This noncanonical stimulation of NFκB triggers the up-regulation of BAG3 and HspB8 expression, two activators of selective autophagy, which relocalize to protein aggregates. Then NFκB-dependent autophagy allows the clearance of protein aggregates. Thus NFκB appears as a central and major regulator of protein aggregate clearance by modulating autophagic activity. In this context, the pharmacological stimulation of this quality control pathway might represent a valuable strategy for therapies against protein conformational diseases.
© 2016 Nivon et al. This article is distributed by The American Society for Cell Biology under license from the author(s). Two months after publication it is available to the public under an Attribution–Noncommercial–Share Alike 3.0 Unported Creative Commons License (http://creativecommons.org/licenses/by-nc-sa/3.0).
Figures
Similar articles
-
Modulation of Protein Quality Control and Proteasome to Autophagy Switch in Immortalized Myoblasts from Duchenne Muscular Dystrophy Patients.Int J Mol Sci. 2018 Jan 7;19(1):178. doi: 10.3390/ijms19010178. Int J Mol Sci. 2018. PMID: 29316663 Free PMC article.
-
NF-κB regulates protein quality control after heat stress through modulation of the BAG3-HspB8 complex.J Cell Sci. 2012 Mar 1;125(Pt 5):1141-51. doi: 10.1242/jcs.091041. Epub 2012 Feb 2. J Cell Sci. 2012. PMID: 22302993
-
Zebrafish models of BAG3 myofibrillar myopathy suggest a toxic gain of function leading to BAG3 insufficiency.Acta Neuropathol. 2014 Dec;128(6):821-33. doi: 10.1007/s00401-014-1344-5. Epub 2014 Oct 2. Acta Neuropathol. 2014. PMID: 25273835
-
HspB8 and Bag3: a new chaperone complex targeting misfolded proteins to macroautophagy.Autophagy. 2008 Feb;4(2):237-9. doi: 10.4161/auto.5407. Epub 2007 Dec 11. Autophagy. 2008. PMID: 18094623 Review.
-
IKK connects autophagy to major stress pathways.Autophagy. 2010 Jan;6(1):189-91. doi: 10.4161/auto.6.1.10818. Autophagy. 2010. PMID: 20110771 Review.
Cited by
-
Neuromuscular Diseases Due to Chaperone Mutations: A Review and Some New Results.Int J Mol Sci. 2020 Feb 19;21(4):1409. doi: 10.3390/ijms21041409. Int J Mol Sci. 2020. PMID: 32093037 Free PMC article. Review.
-
Modulation of Protein Quality Control and Proteasome to Autophagy Switch in Immortalized Myoblasts from Duchenne Muscular Dystrophy Patients.Int J Mol Sci. 2018 Jan 7;19(1):178. doi: 10.3390/ijms19010178. Int J Mol Sci. 2018. PMID: 29316663 Free PMC article.
-
The Role of HSPB8, a Component of the Chaperone-Assisted Selective Autophagy Machinery, in Cancer.Cells. 2021 Feb 5;10(2):335. doi: 10.3390/cells10020335. Cells. 2021. PMID: 33562660 Free PMC article. Review.
-
The Endoplasmic Reticulum Stress Response in Neuroprogressive Diseases: Emerging Pathophysiological Role and Translational Implications.Mol Neurobiol. 2018 Dec;55(12):8765-8787. doi: 10.1007/s12035-018-1028-6. Epub 2018 Mar 29. Mol Neurobiol. 2018. PMID: 29594942 Free PMC article. Review.
-
Boosting mTOR-dependent autophagy via upstream TLR4-MyD88-MAPK signalling and downstream NF-κB pathway quenches intestinal inflammation and oxidative stress injury.EBioMedicine. 2018 Sep;35:345-360. doi: 10.1016/j.ebiom.2018.08.035. Epub 2018 Aug 29. EBioMedicine. 2018. PMID: 30170968 Free PMC article.
References
-
- Bampton ET, Goemans CG, Niranjan D, Mizushima N, Tolkovsky AM. The dynamics of autophagy visualized in live cells: from autophagosome formation to fusion with endo/lysosomes. Autophagy. 2005;1:23–36. - PubMed
-
- Carra S. The stress-inducible HspB8-Bag3 complex induces the eIF2alpha kinase pathway: implications for protein quality control and viral factory degradation. Autophagy. 2009;5:428–429. - PubMed
-
- Carra S, Seguin SJ, Lambert H, Landry J. HspB8 chaperone activity toward poly(Q)-containing proteins depends on its association with Bag3, a stimulator of macroautophagy. J Biol Chem. 2008a;283:1437–1444. - PubMed
-
- Carra S, Seguin SJ, Landry J. HspB8 and Bag3: a new chaperone complex targeting misfolded proteins to macroautophagy. Autophagy. 2008b;4:237–239. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
