

6-Mercaptopurine attenuates tumor necrosis factor-α production in microglia through Nur77-mediated transrepression and PI3K/Akt/mTOR signaling-mediated translational regulation
- PMID: 27075886
- PMCID: PMC4831152
- DOI: 10.1186/s12974-016-0543-5
6-Mercaptopurine attenuates tumor necrosis factor-α production in microglia through Nur77-mediated transrepression and PI3K/Akt/mTOR signaling-mediated translational regulation
Abstract
Background: The pathogenesis of several neurodegenerative diseases often involves the microglial activation and associated inflammatory processes. Activated microglia release pro-inflammatory factors that may be neurotoxic. 6-Mercaptopurine (6-MP) is a well-established immunosuppressive drug. Common understanding of their immunosuppressive properties is largely limited to peripheral immune cells. However, the effect of 6-MP in the central nervous system, especially in microglia in the context of neuroinflammation is, as yet, unclear. Tumor necrosis factor-α (TNF-α) is a key cytokine of the immune system that initiates and promotes neuroinflammation. The present study aimed to investigate the effect of 6-MP on TNF-α production by microglia to discern the molecular mechanisms of this modulation.
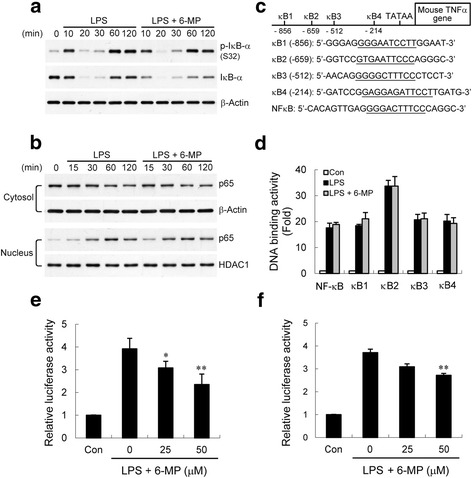
Methods: Lipopolysaccharide (LPS) was used to induce an inflammatory response in cultured primary microglia or murine BV-2 microglial cells. Released TNF-α was measured by enzyme-linked immunosorbent assay (ELISA). Gene expression was determined by real-time reverse transcription polymerase chain reaction (RT-PCR). Signaling molecules were analyzed by western blotting, and activation of NF-κB was measured by ELISA-based DNA binding analysis and luciferase reporter assay. Chromatin immunoprecipitation (ChIP) analysis was performed to examine NF-κB p65 and coactivator p300 enrichments and histone modifications at the endogenous TNF-α promoter.
Results: Treatment of LPS-activated microglia with 6-MP significantly attenuated TNF-α production. In 6-MP pretreated microglia, LPS-induced MAPK signaling, IκB-α degradation, NF-κB p65 nuclear translocation, and in vitro p65 DNA binding activity were not impaired. However, 6-MP suppressed transactivation activity of NF-κB and TNF-α promoter by inhibiting phosphorylation and acetylation of p65 on Ser276 and Lys310, respectively. ChIP analyses revealed that 6-MP dampened LPS-induced histone H3 acetylation of chromatin surrounding the TNF-α promoter, ultimately leading to a decrease in p65/coactivator-mediated transcription of TNF-α gene. Furthermore, 6-MP enhanced orphan nuclear receptor Nur77 expression. Using RNA interference approach, we further demonstrated that Nur77 upregulation contribute to 6-MP-mediated inhibitory effect on TNF-α production. Additionally, 6-MP also impeded TNF-α mRNA translation through prevention of LPS-activated PI3K/Akt/mTOR signaling cascades.
Conclusions: These results suggest that 6-MP might have a therapeutic potential in neuroinflammation-related neurodegenerative disorders through downregulation of microglia-mediated inflammatory processes.
Keywords: 6-Mercaptopurine; Histone H3 acetylation; Microglia; Nuclear factor-κB; Nur77; PI3K/Akt; TNF-α; mTOR.
Figures










References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
