

Computational protein-ligand docking and virtual drug screening with the AutoDock suite
- PMID: 27077332
- PMCID: PMC4868550
- DOI: 10.1038/nprot.2016.051
Computational protein-ligand docking and virtual drug screening with the AutoDock suite
Abstract
Computational docking can be used to predict bound conformations and free energies of binding for small-molecule ligands to macromolecular targets. Docking is widely used for the study of biomolecular interactions and mechanisms, and it is applied to structure-based drug design. The methods are fast enough to allow virtual screening of ligand libraries containing tens of thousands of compounds. This protocol covers the docking and virtual screening methods provided by the AutoDock suite of programs, including a basic docking of a drug molecule with an anticancer target, a virtual screen of this target with a small ligand library, docking with selective receptor flexibility, active site prediction and docking with explicit hydration. The entire protocol will require ∼5 h.
Conflict of interest statement
The authors declare no competing financial interests
Figures
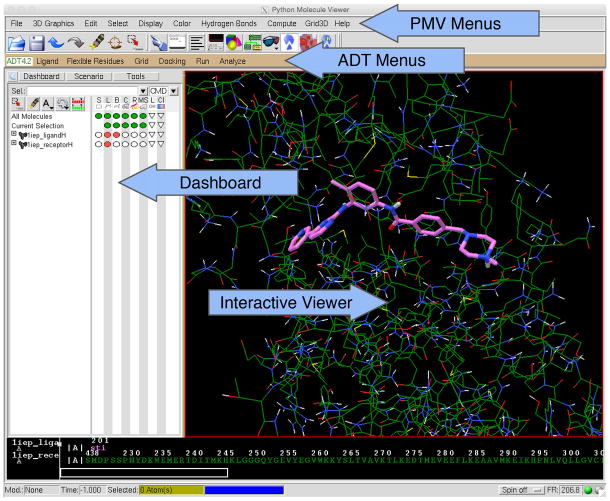
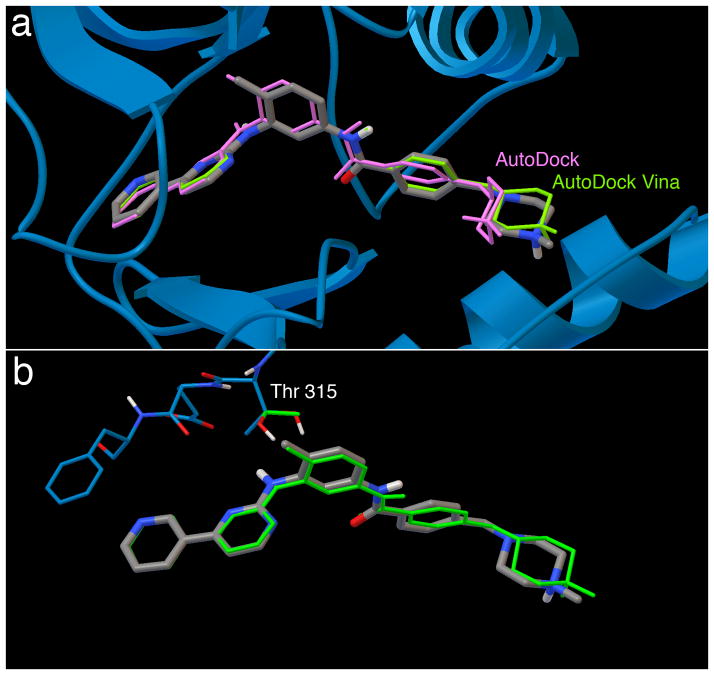
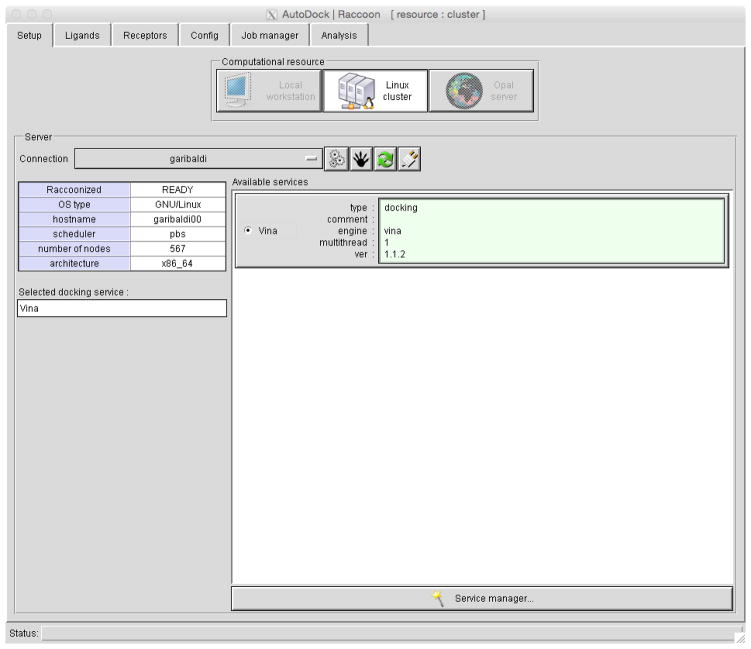
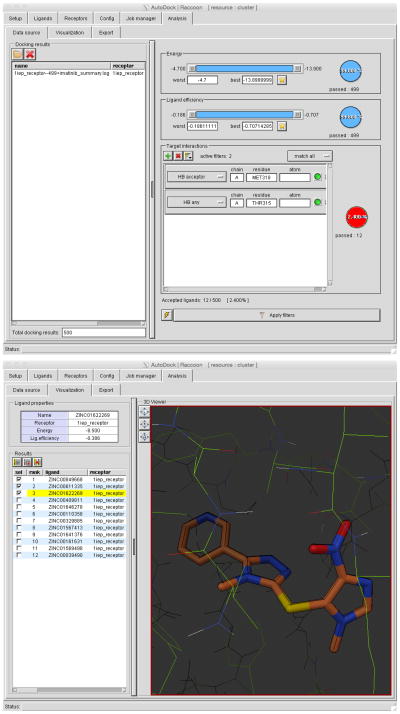
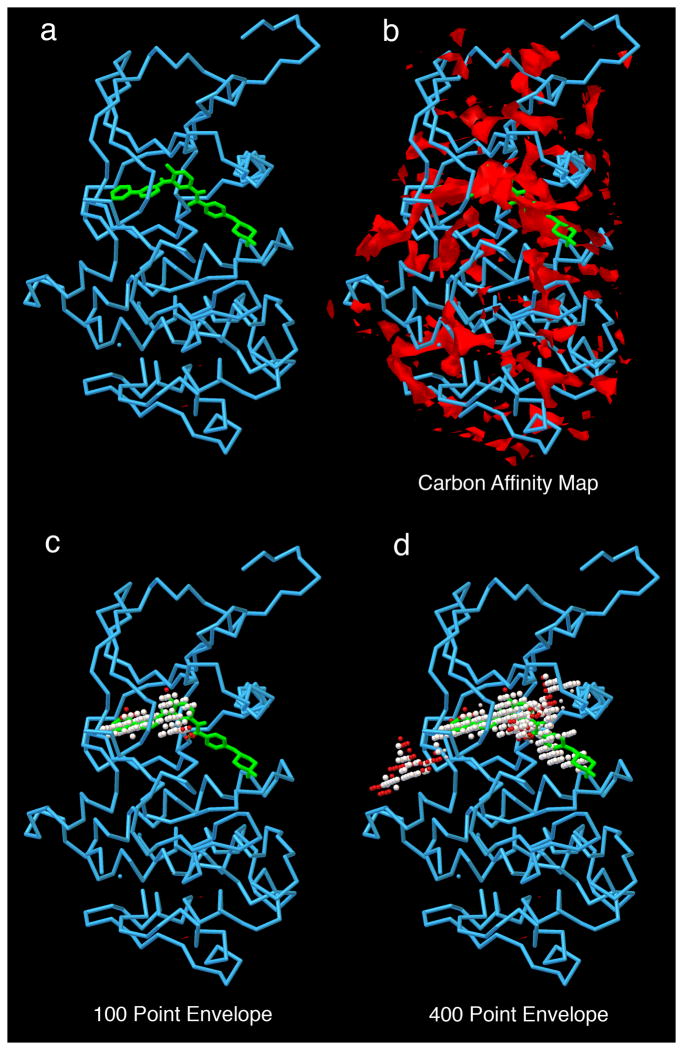
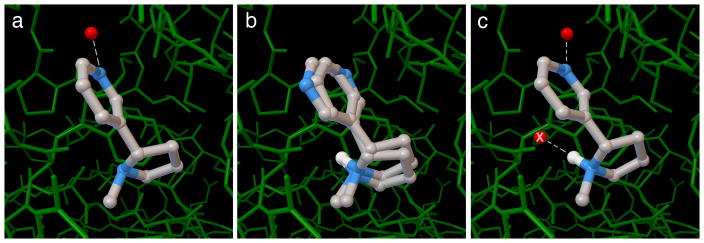
References
-
- Goodsell DS, Olson AJ. Automated docking of substrates to proteins by simulated annealing. Proteins: Struct Funct Genet. 1990;8:195–202. - PubMed
-
- Morris GM, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comp Chem. 1998;19:1639–1662.
-
- Forli Raccoon for Processing Virtual Screening. 2013 < http://autodock.scripps.edu/resources/raccoon>.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
