

Translocation and deletion breakpoints in cancer genomes are associated with potential non-B DNA-forming sequences
- PMID: 27084947
- PMCID: PMC4937311
- DOI: 10.1093/nar/gkw261
Translocation and deletion breakpoints in cancer genomes are associated with potential non-B DNA-forming sequences
Abstract
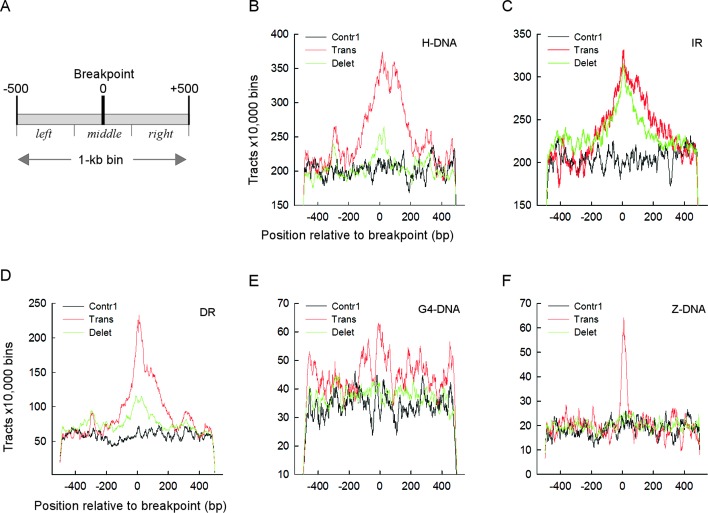
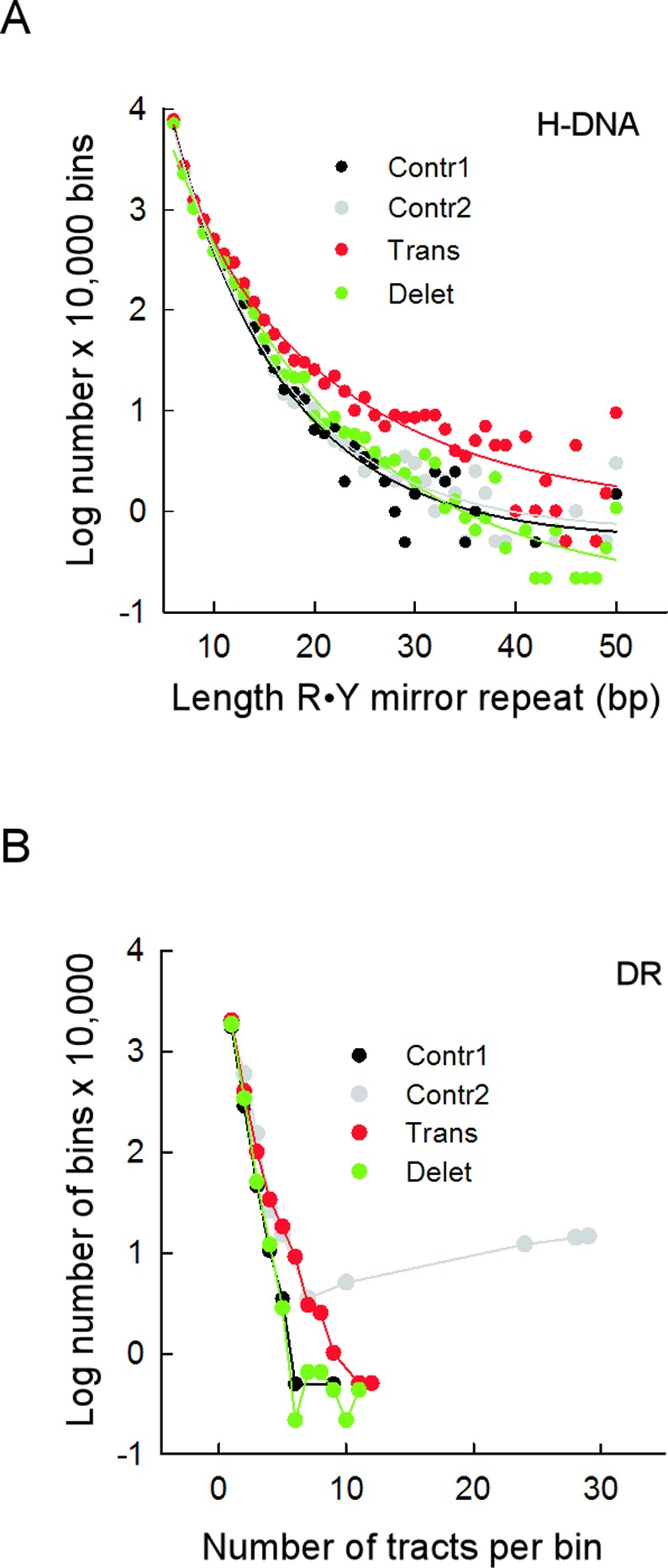
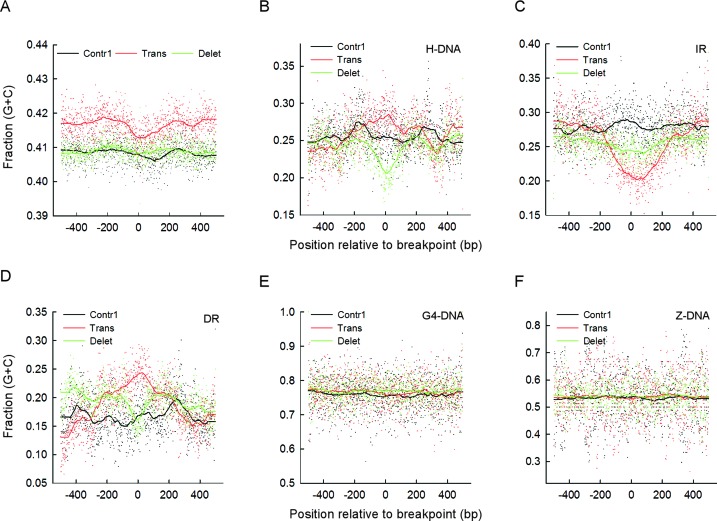
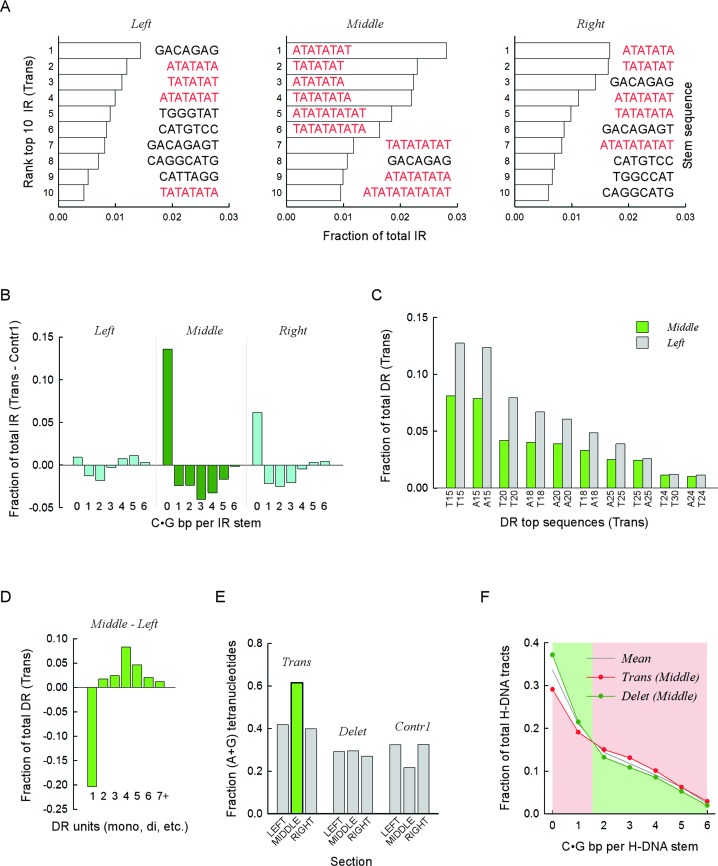
Gross chromosomal rearrangements (including translocations, deletions, insertions and duplications) are a hallmark of cancer genomes and often create oncogenic fusion genes. An obligate step in the generation of such gross rearrangements is the formation of DNA double-strand breaks (DSBs). Since the genomic distribution of rearrangement breakpoints is non-random, intrinsic cellular factors may predispose certain genomic regions to breakage. Notably, certain DNA sequences with the potential to fold into secondary structures [potential non-B DNA structures (PONDS); e.g. triplexes, quadruplexes, hairpin/cruciforms, Z-DNA and single-stranded looped-out structures with implications in DNA replication and transcription] can stimulate the formation of DNA DSBs. Here, we tested the postulate that these DNA sequences might be found at, or in close proximity to, rearrangement breakpoints. By analyzing the distribution of PONDS-forming sequences within ±500 bases of 19 947 translocation and 46 365 sequence-characterized deletion breakpoints in cancer genomes, we find significant association between PONDS-forming repeats and cancer breakpoints. Specifically, (AT)n, (GAA)n and (GAAA)n constitute the most frequent repeats at translocation breakpoints, whereas A-tracts occur preferentially at deletion breakpoints. Translocation breakpoints near PONDS-forming repeats also recur in different individuals and patient tumor samples. Hence, PONDS-forming sequences represent an intrinsic risk factor for genomic rearrangements in cancer genomes.
© The Author(s) 2016. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Mertens F., Johansson B., Fioretos T., Mitelman F. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer. 2015;15:371–381. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
