

De Novo Mutations of RERE Cause a Genetic Syndrome with Features that Overlap Those Associated with Proximal 1p36 Deletions
- PMID: 27087320
- PMCID: PMC4863473
- DOI: 10.1016/j.ajhg.2016.03.002
De Novo Mutations of RERE Cause a Genetic Syndrome with Features that Overlap Those Associated with Proximal 1p36 Deletions
Abstract
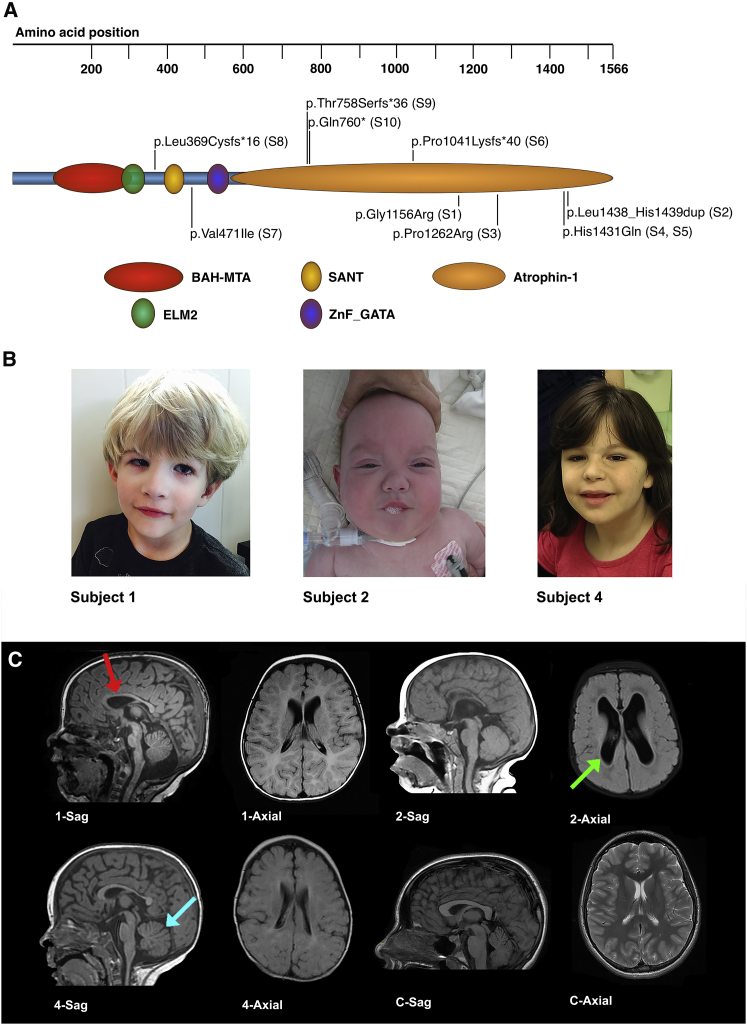
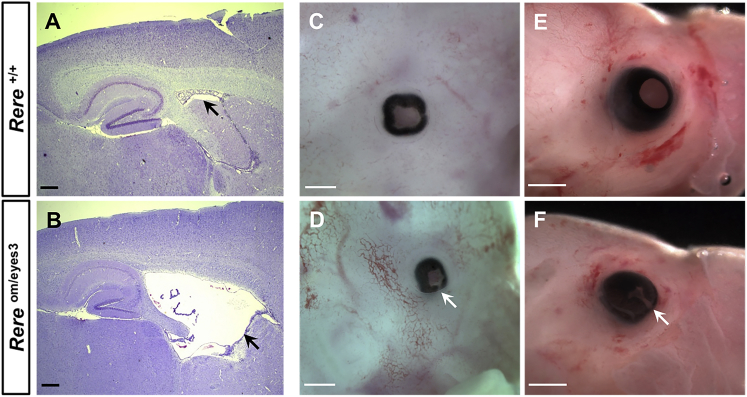
Deletions of chromosome 1p36 affect approximately 1 in 5,000 newborns and are associated with developmental delay, intellectual disability, and defects involving the brain, eye, ear, heart, and kidney. Arginine-glutamic acid dipeptide repeats (RERE) is located in the proximal 1p36 critical region. RERE is a widely-expressed nuclear receptor coregulator that positively regulates retinoic acid signaling. Animal models suggest that RERE deficiency might contribute to many of the structural and developmental birth defects and medical problems seen in individuals with 1p36 deletion syndrome, although human evidence supporting this role has been lacking. In this report, we describe ten individuals with intellectual disability, developmental delay, and/or autism spectrum disorder who carry rare and putatively damaging changes in RERE. In all cases in which both parental DNA samples were available, these changes were found to be de novo. Associated features that were recurrently seen in these individuals included hypotonia, seizures, behavioral problems, structural CNS anomalies, ophthalmologic anomalies, congenital heart defects, and genitourinary abnormalities. The spectrum of defects documented in these individuals is similar to that of a cohort of 31 individuals with isolated 1p36 deletions that include RERE and are recapitulated in RERE-deficient zebrafish and mice. Taken together, our findings suggest that mutations in RERE cause a genetic syndrome and that haploinsufficiency of RERE might be sufficient to cause many of the phenotypes associated with proximal 1p36 deletions.
Copyright © 2016 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Shaffer L.G., Lupski J.R. Molecular mechanisms for constitutional chromosomal rearrangements in humans. Annu. Rev. Genet. 2000;34:297–329. - PubMed
-
- Heilstedt H.A., Ballif B.C., Howard L.A., Kashork C.D., Shaffer L.G. Population data suggest that deletions of 1p36 are a relatively common chromosome abnormality. Clin. Genet. 2003;64:310–316. - PubMed
-
- Giraudeau F., Taine L., Biancalana V., Delobel B., Journel H., Missirian C., Lacombe D., Bonneau D., Parent P., Aubert D. Use of a set of highly polymorphic minisatellite probes for the identification of cryptic 1p36.3 deletions in a large collection of patients with idiopathic mental retardation. J. Med. Genet. 2001;38:121–125. - PMC - PubMed
-
- Battaglia A., Hoyme H.E., Dallapiccola B., Zackai E., Hudgins L., McDonald-McGinn D., Bahi-Buisson N., Romano C., Williams C.A., Brailey L.L. Further delineation of deletion 1p36 syndrome in 60 patients: a recognizable phenotype and common cause of developmental delay and mental retardation. Pediatrics. 2008;121:404–410. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
