

Oncogenic KRAS Regulates Tumor Cell Signaling via Stromal Reciprocation
- PMID: 27087446
- PMCID: PMC4868820
- DOI: 10.1016/j.cell.2016.03.029
Oncogenic KRAS Regulates Tumor Cell Signaling via Stromal Reciprocation
Erratum in
-
Oncogenic KRAS Regulates Tumor Cell Signaling via Stromal Reciprocation.Cell. 2016 Jun 16;165(7):1818. doi: 10.1016/j.cell.2016.05.079. Cell. 2016. PMID: 27315484 Free PMC article. No abstract available.
Abstract
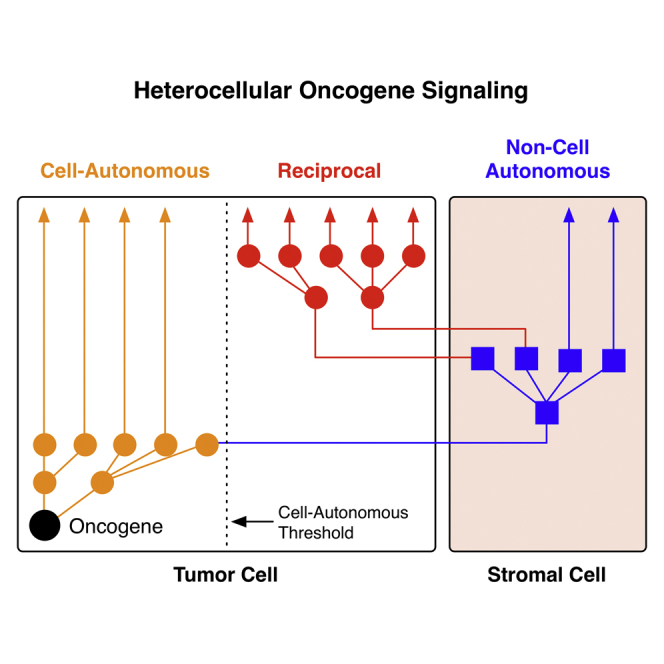
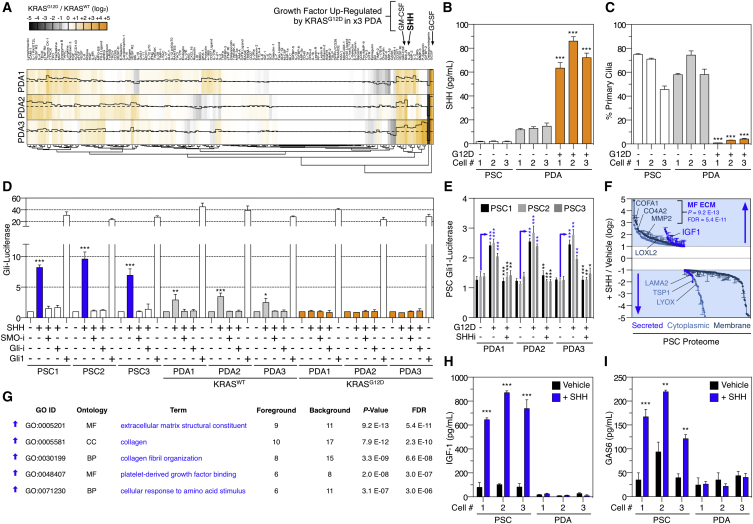
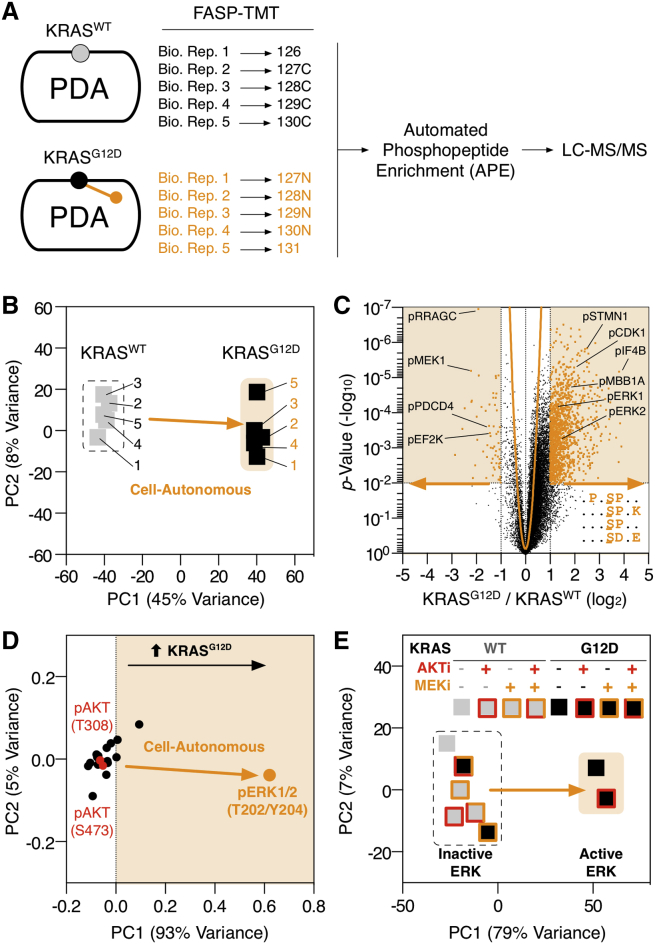
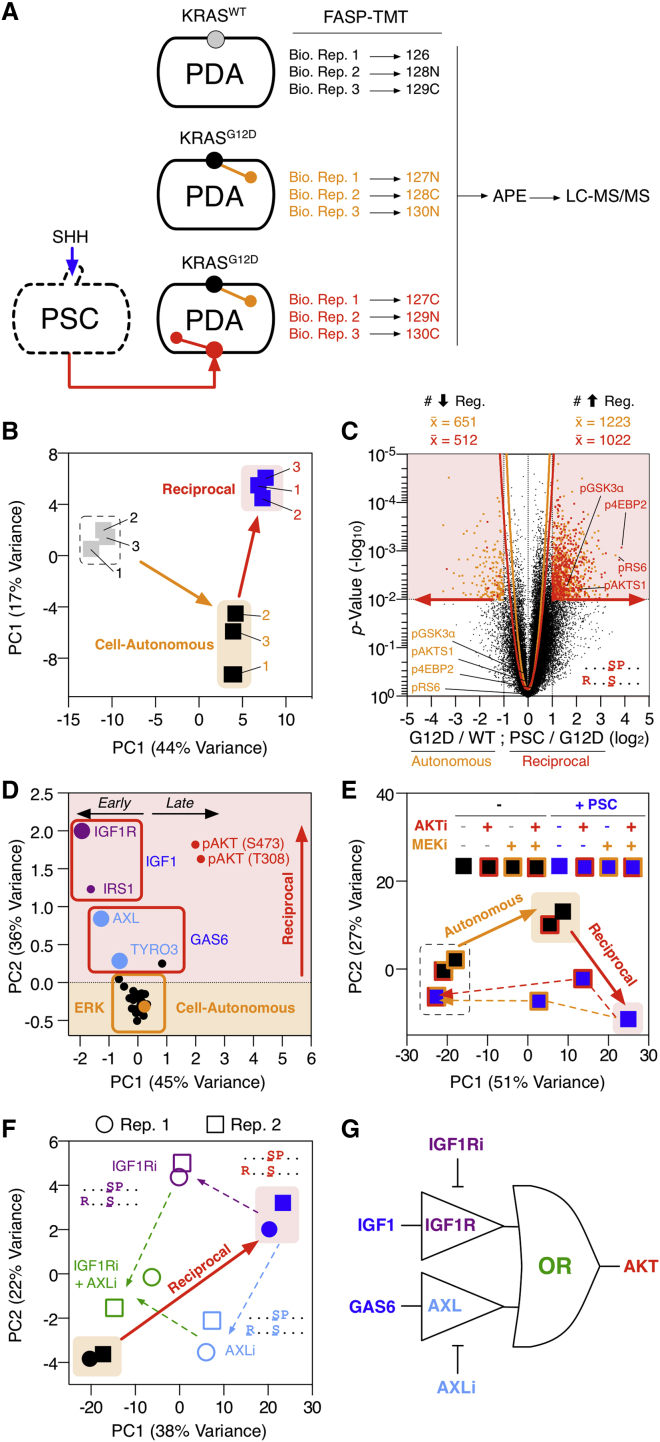
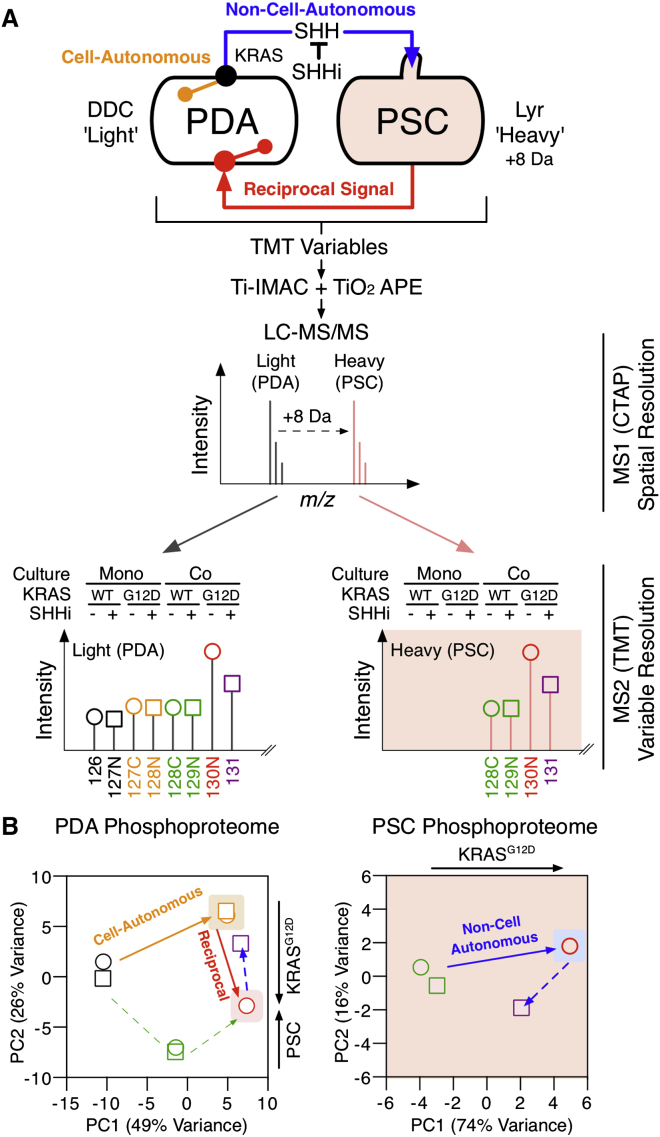
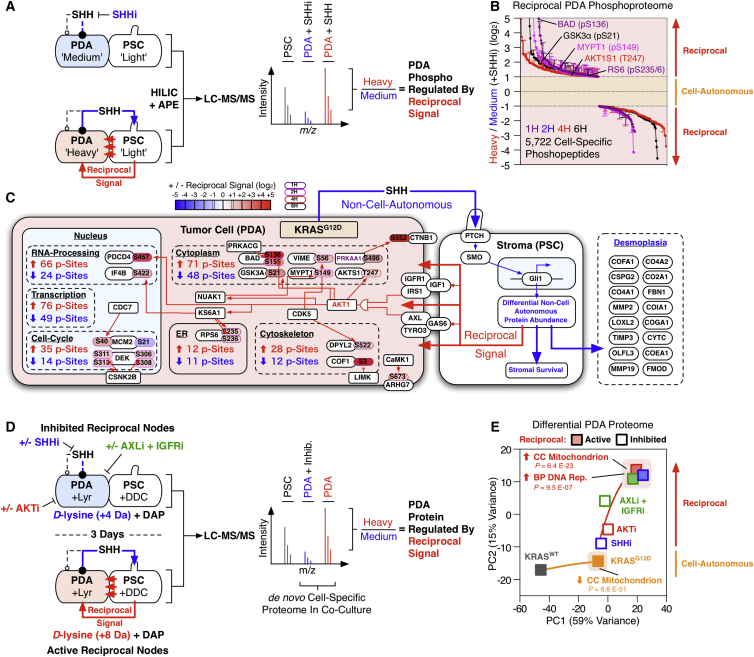
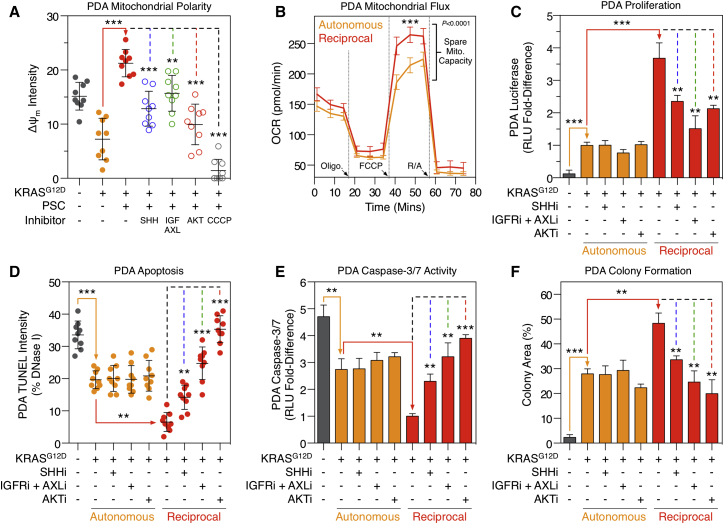
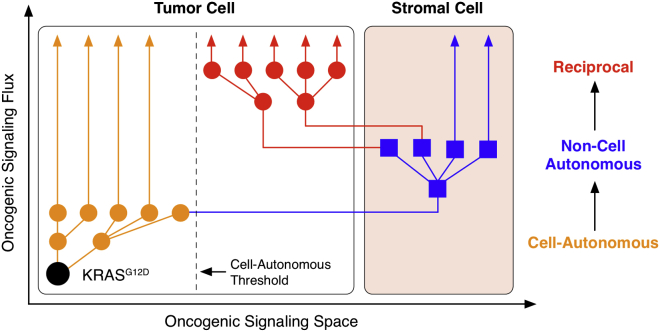
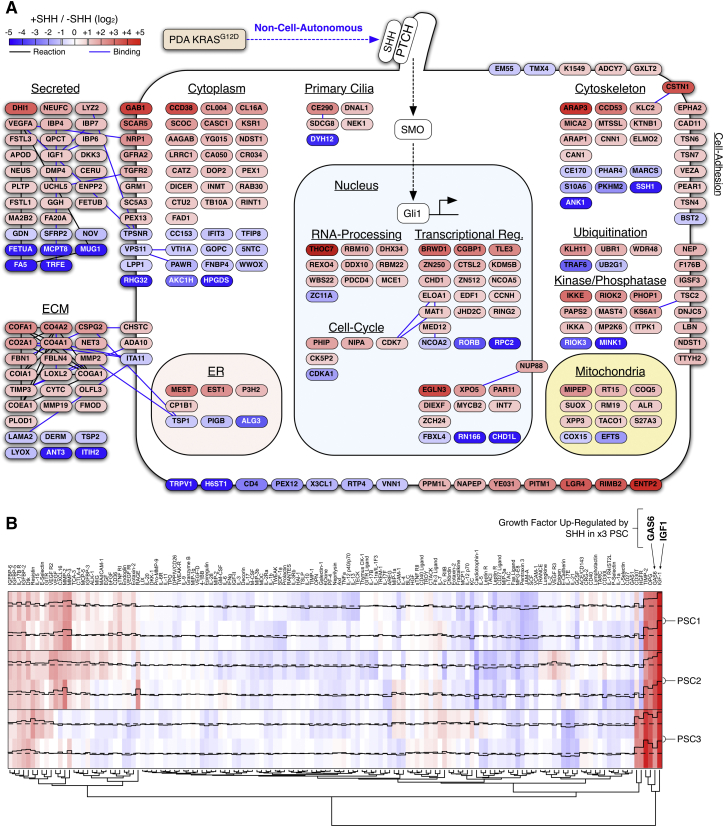
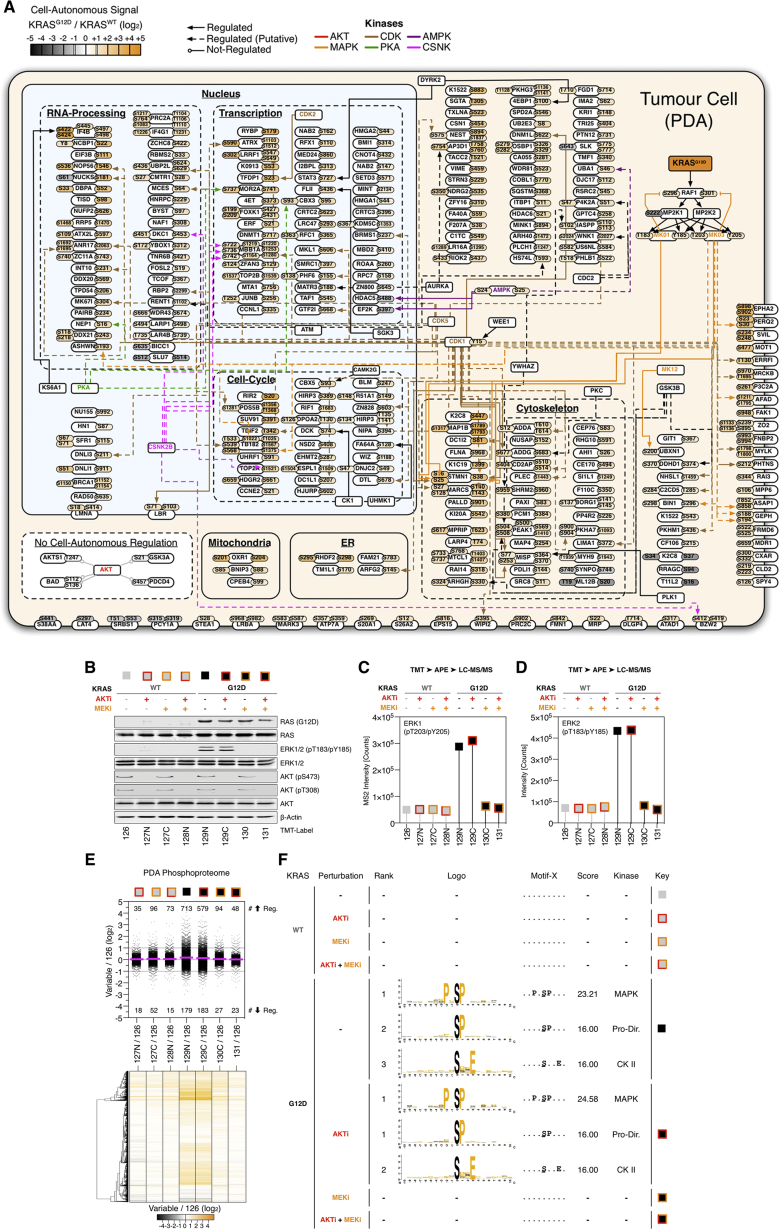
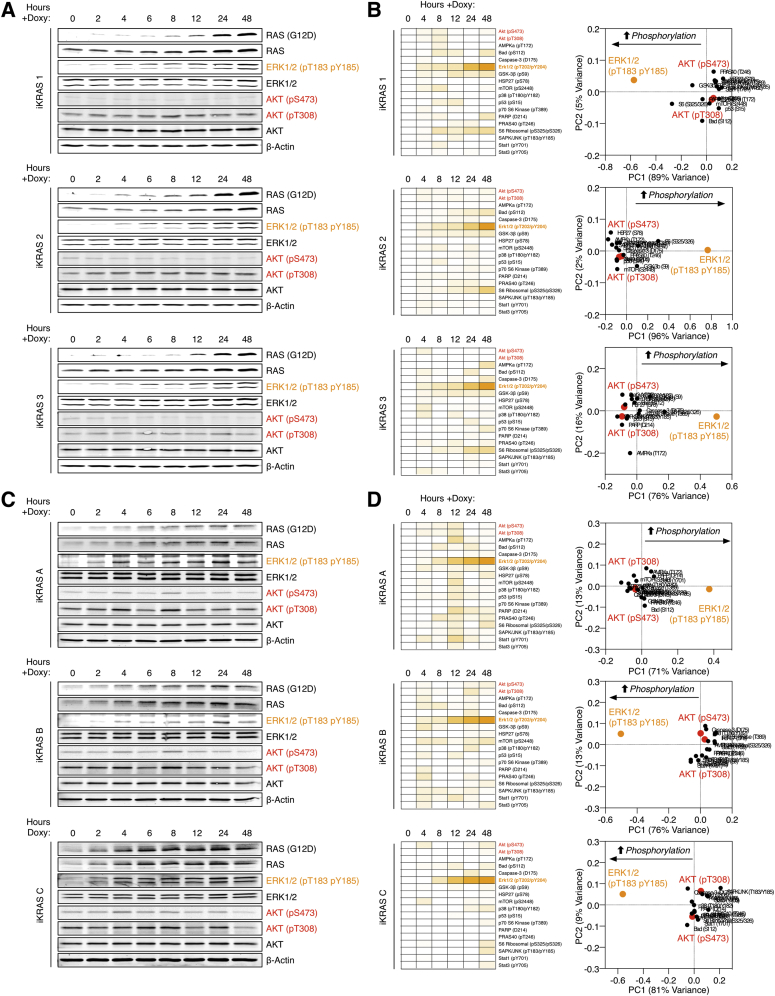
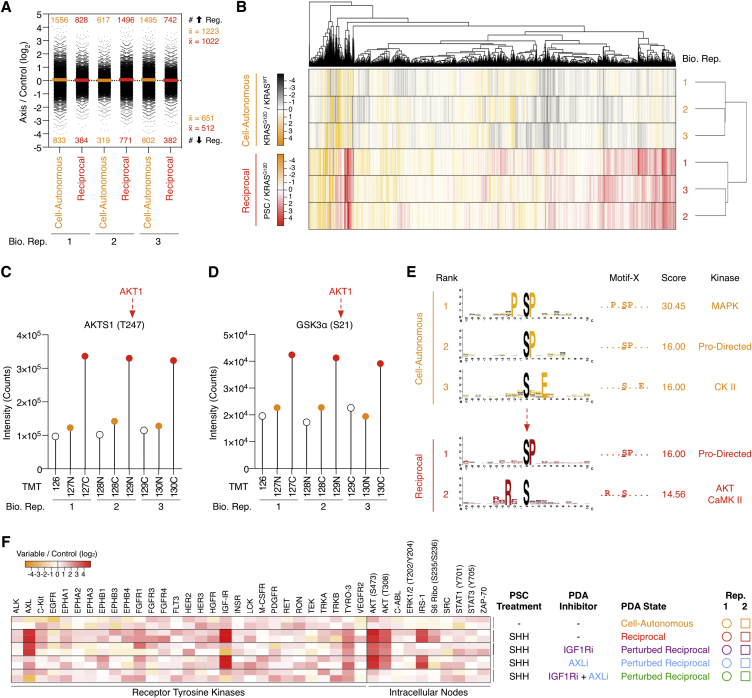
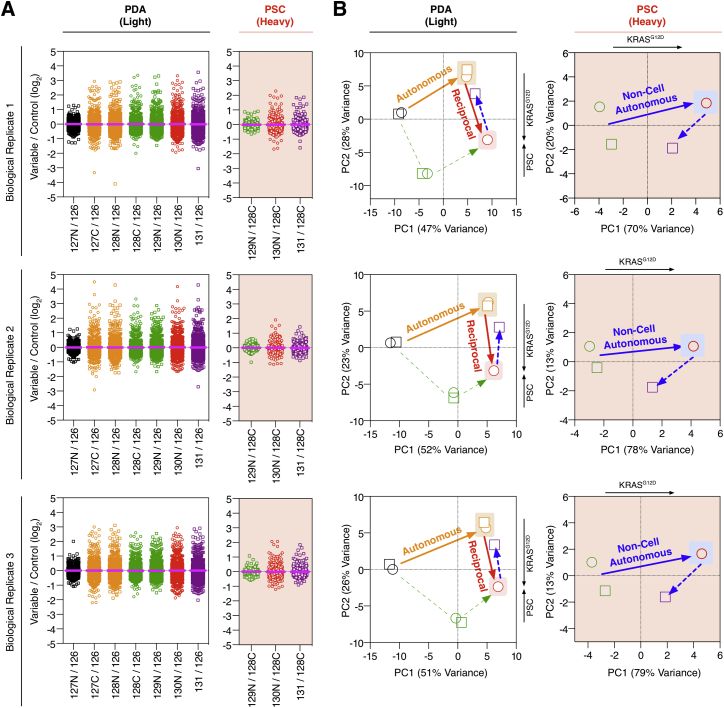
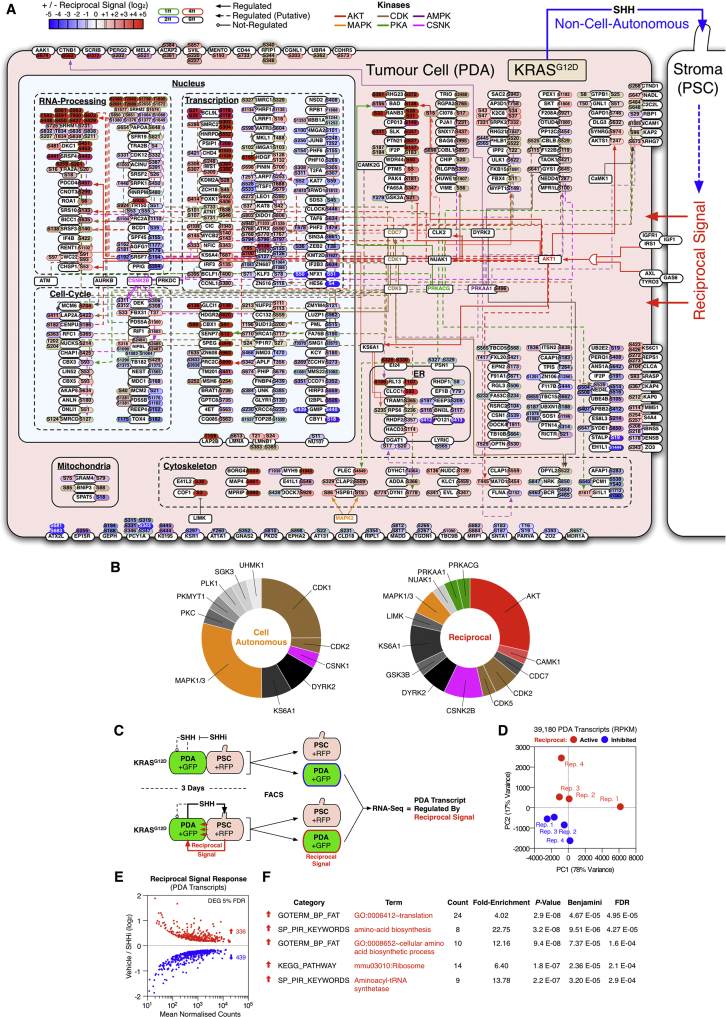
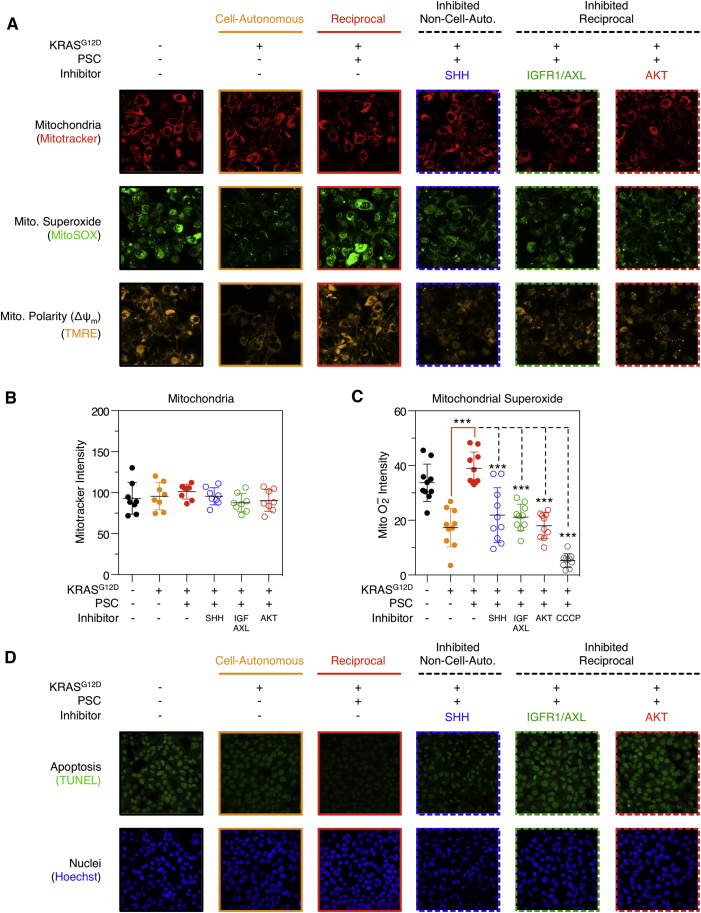
Oncogenic mutations regulate signaling within both tumor cells and adjacent stromal cells. Here, we show that oncogenic KRAS (KRAS(G12D)) also regulates tumor cell signaling via stromal cells. By combining cell-specific proteome labeling with multivariate phosphoproteomics, we analyzed heterocellular KRAS(G12D) signaling in pancreatic ductal adenocarcinoma (PDA) cells. Tumor cell KRAS(G12D) engages heterotypic fibroblasts, which subsequently instigate reciprocal signaling in the tumor cells. Reciprocal signaling employs additional kinases and doubles the number of regulated signaling nodes from cell-autonomous KRAS(G12D). Consequently, reciprocal KRAS(G12D) produces a tumor cell phosphoproteome and total proteome that is distinct from cell-autonomous KRAS(G12D) alone. Reciprocal signaling regulates tumor cell proliferation and apoptosis and increases mitochondrial capacity via an IGF1R/AXL-AKT axis. These results demonstrate that oncogene signaling should be viewed as a heterocellular process and that our existing cell-autonomous perspective underrepresents the extent of oncogene signaling in cancer. VIDEO ABSTRACT.
Copyright © 2016 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Almoguera C., Shibata D., Forrester K., Martin J., Arnheim N., Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–554. - PubMed
-
- Baer R., Cintas C., Dufresne M., Cassant-Sourdy S., Schönhuber N., Planque L., Lulka H., Couderc B., Bousquet C., Garmy-Susini B. Pancreatic cell plasticity and cancer initiation induced by oncogenic Kras is completely dependent on wild-type PI 3-kinase p110α. Genes Dev. 2014;28:2621–2635. - PMC - PubMed
-
- Brock A., Krause S., Ingber D.E. Control of cancer formation by intrinsic genetic noise and microenvironmental cues. Nat. Rev. Cancer. 2015;15:499–509. - PubMed
-
- Croce C.M. Oncogenes and cancer. N. Engl. J. Med. 2008;358:502–511. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
