

Pooled ChIP-Seq Links Variation in Transcription Factor Binding to Complex Disease Risk
- PMID: 27087447
- PMCID: PMC4842172
- DOI: 10.1016/j.cell.2016.03.041
Pooled ChIP-Seq Links Variation in Transcription Factor Binding to Complex Disease Risk
Abstract
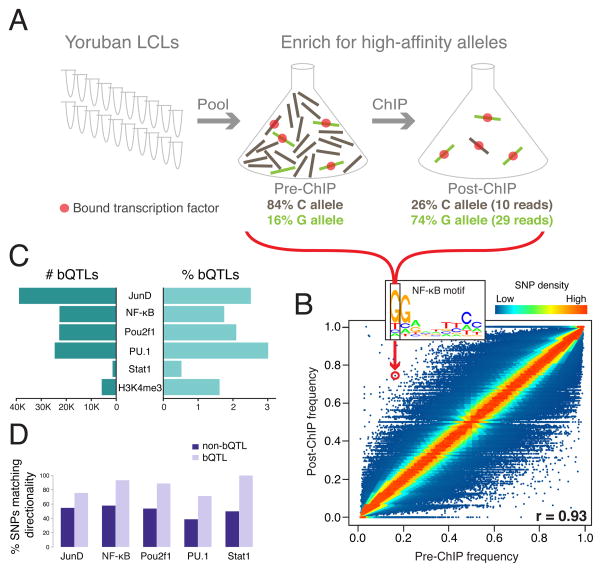
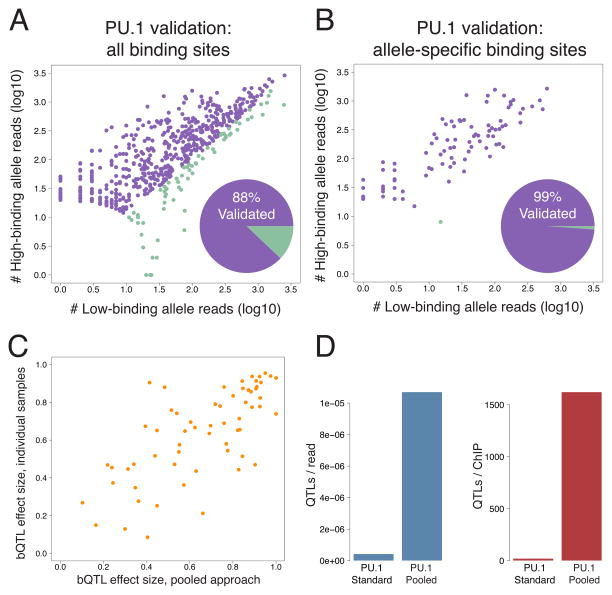
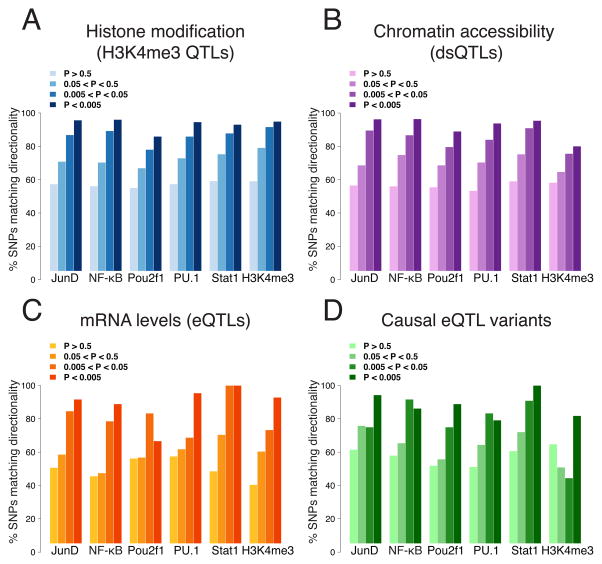
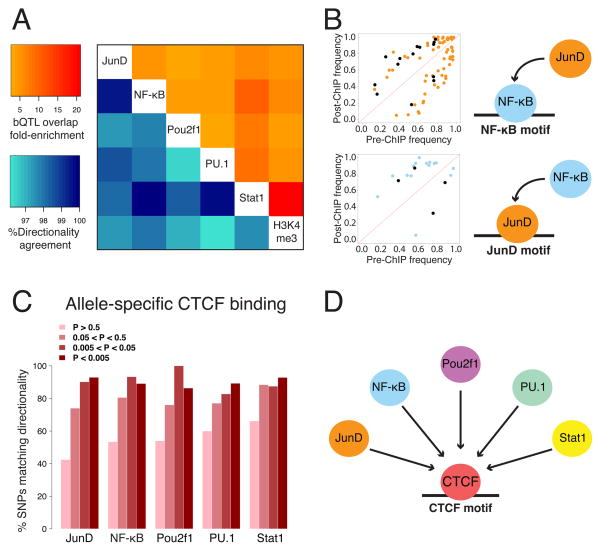
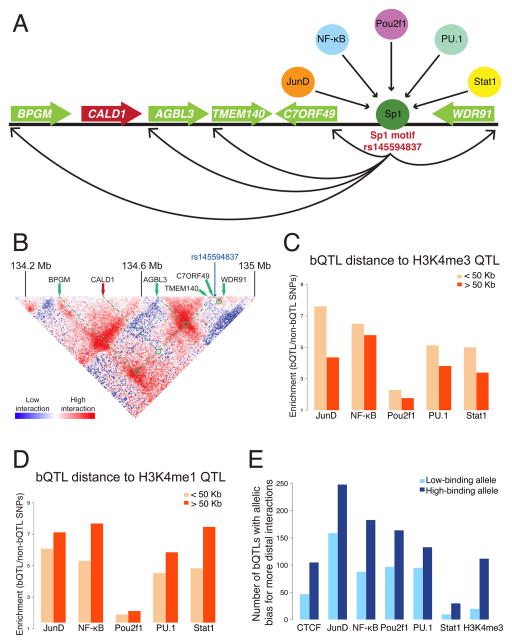
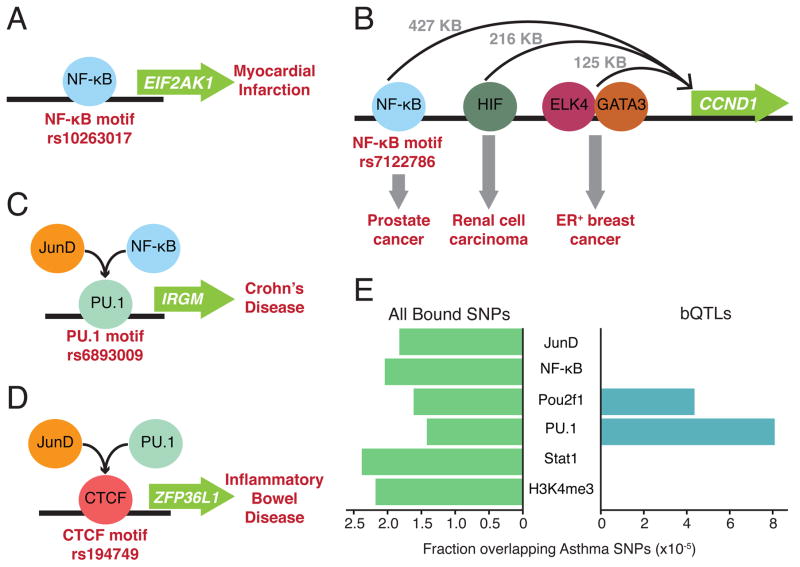
Cis-regulatory elements such as transcription factor (TF) binding sites can be identified genome-wide, but it remains far more challenging to pinpoint genetic variants affecting TF binding. Here, we introduce a pooling-based approach to mapping quantitative trait loci (QTLs) for molecular-level traits. Applying this to five TFs and a histone modification, we mapped thousands of cis-acting QTLs, with over 25-fold lower cost compared to standard QTL mapping. We found that single genetic variants frequently affect binding of multiple TFs, and CTCF can recruit all five TFs to its binding sites. These QTLs often affect local chromatin and transcription but can also influence long-range chromosomal contacts, demonstrating a role for natural genetic variation in chromosomal architecture. Thousands of these QTLs have been implicated in genome-wide association studies, providing candidate molecular mechanisms for many disease risk loci and suggesting that TF binding variation may underlie a large fraction of human phenotypic variation.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures
Comment in
-
Genetic variation: Linking TF binding to disease risk using pooled ChIP-seq.Nat Rev Genet. 2016 Jun;17(6):317. doi: 10.1038/nrg.2016.55. Epub 2016 May 3. Nat Rev Genet. 2016. PMID: 27140281 No abstract available.
-
Fishing for Function in the Human Gene Pool.Trends Genet. 2016 Jul;32(7):392-394. doi: 10.1016/j.tig.2016.05.002. Epub 2016 May 21. Trends Genet. 2016. PMID: 27220646 Free PMC article.
References
-
- Aittomäki S, Yang J, Scott EW, Simon MC, Silvennoinen O. Molecular basis of Stat1 and PU.1 cooperation in cytokine-induced Fcgamma receptor I promoter activation. Int Immunol. 2004;16:265–74. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
