

Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus
- PMID: 27091841
- PMCID: PMC4854735
- DOI: 10.1084/jem.20151876
Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus
Abstract
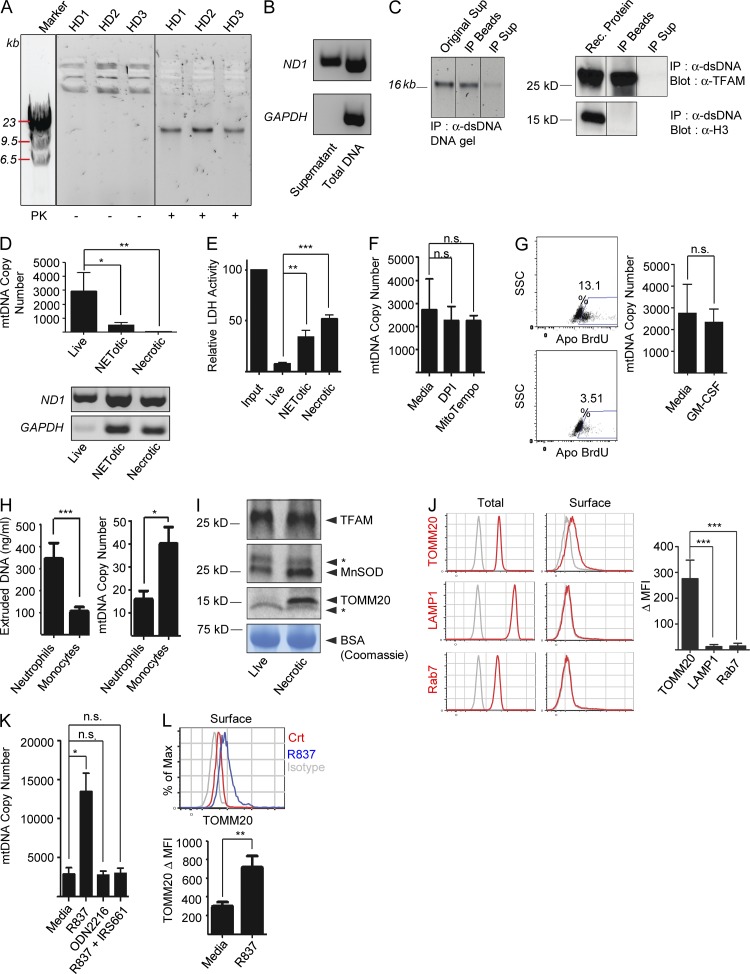
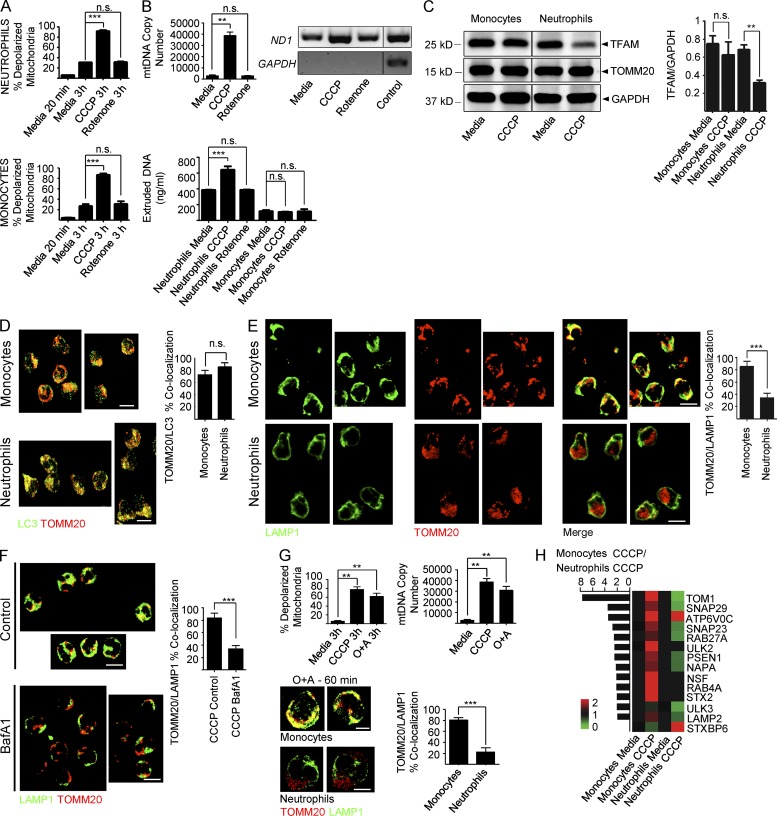
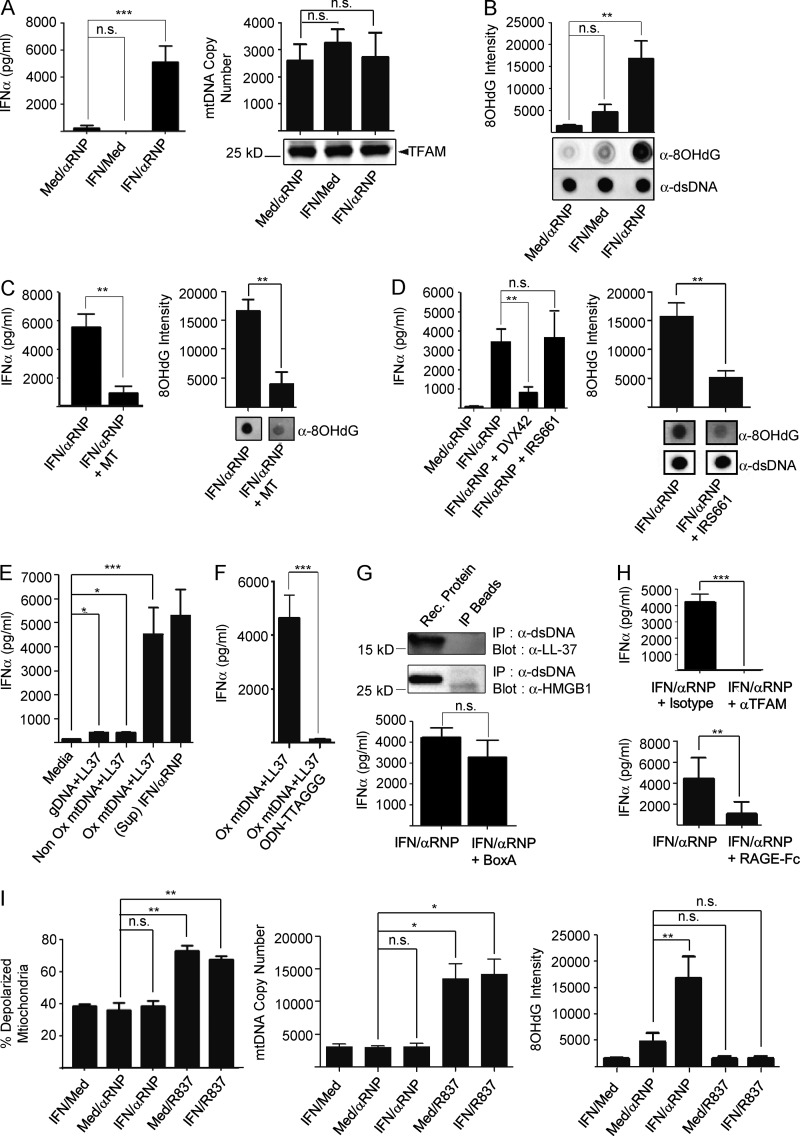
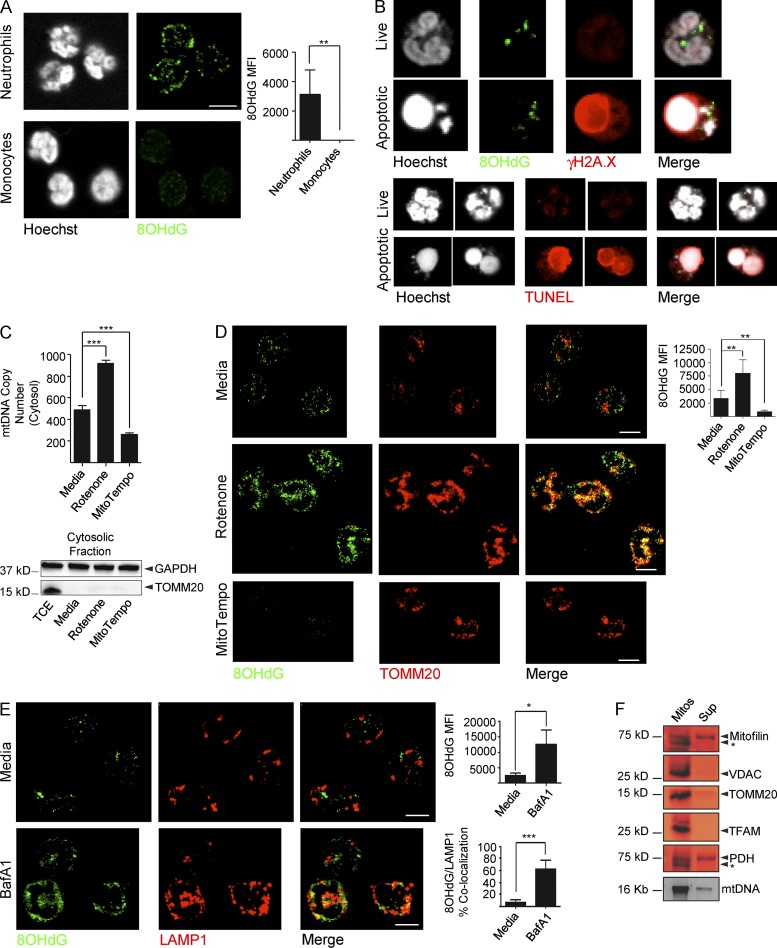
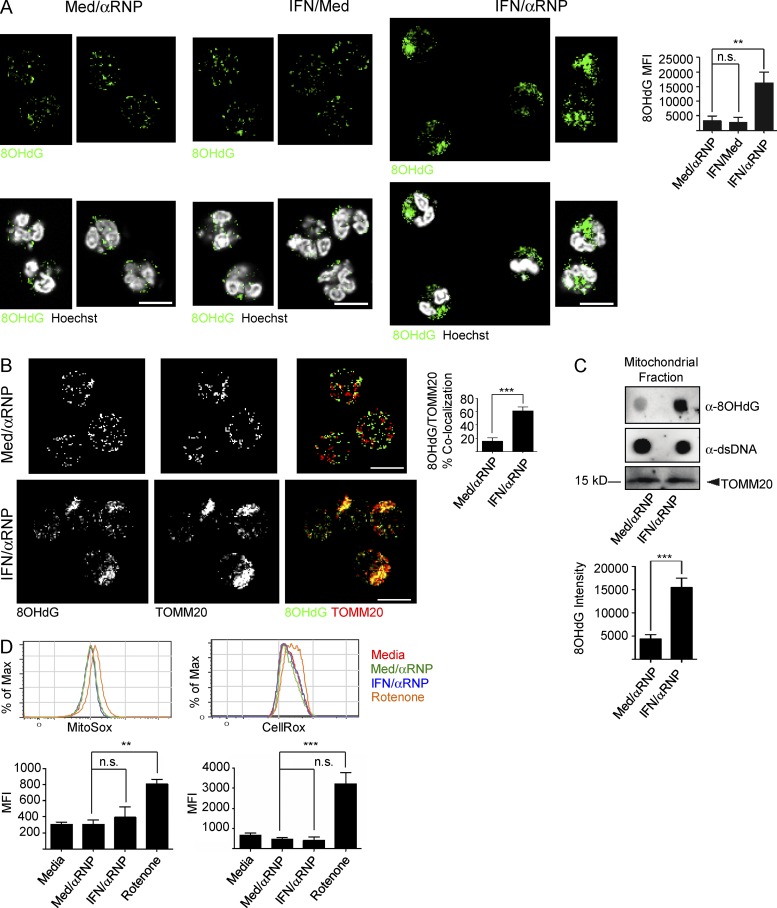
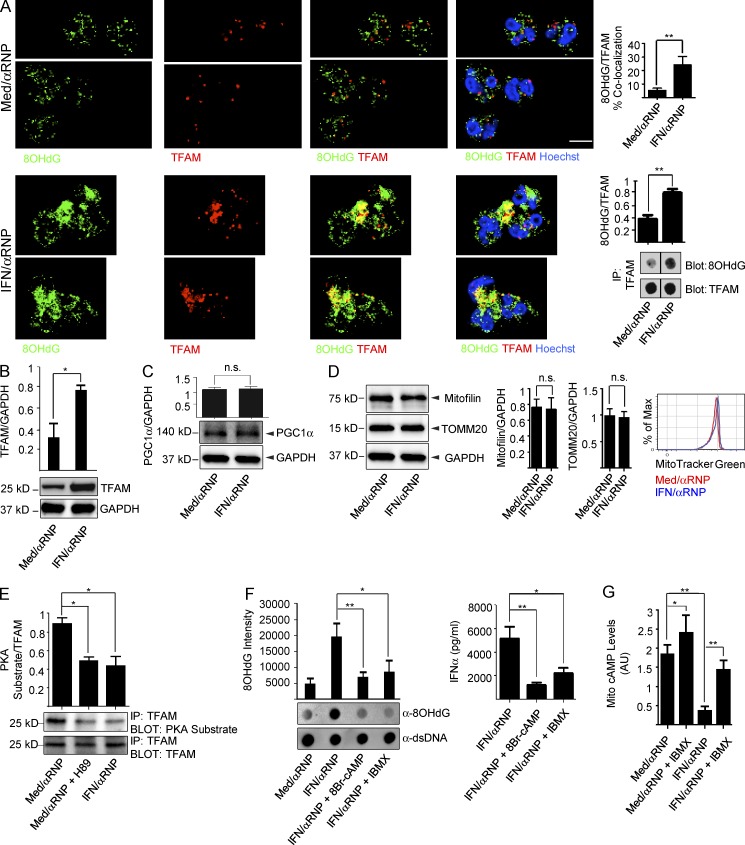
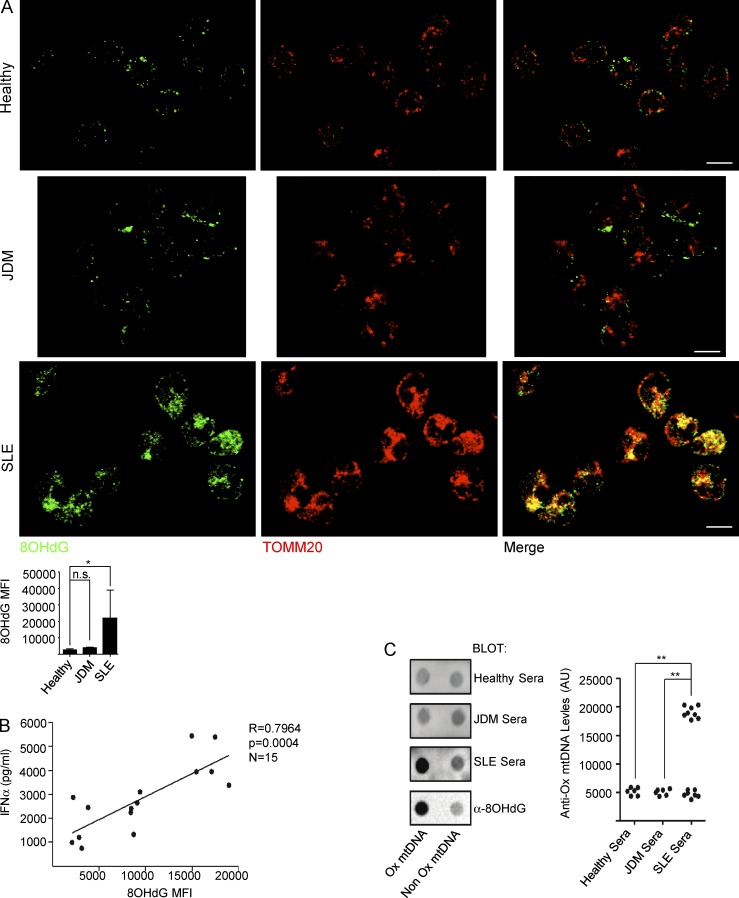
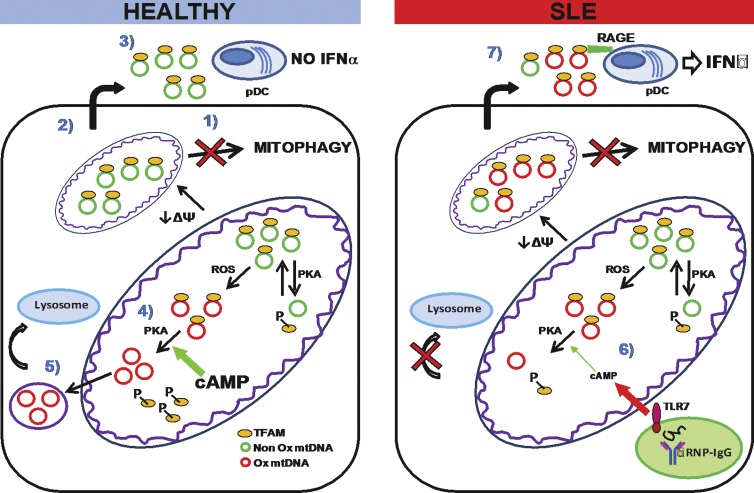
Autoantibodies against nucleic acids and excessive type I interferon (IFN) are hallmarks of human systemic lupus erythematosus (SLE). We previously reported that SLE neutrophils exposed to TLR7 agonist autoantibodies release interferogenic DNA, which we now demonstrate to be of mitochondrial origin. We further show that healthy human neutrophils do not complete mitophagy upon induction of mitochondrial damage. Rather, they extrude mitochondrial components, including DNA (mtDNA), devoid of oxidized (Ox) residues. When mtDNA undergoes oxidation, it is directly routed to lysosomes for degradation. This rerouting requires dissociation from the transcription factor A mitochondria (TFAM), a dual high-mobility group (HMG) protein involved in maintenance and compaction of the mitochondrial genome into nucleoids. Exposure of SLE neutrophils, or healthy IFN-primed neutrophils, to antiribonucleotide protein autoantibodies blocks TFAM phosphorylation, a necessary step for nucleoid dissociation. Consequently, Ox nucleoids accumulate within mitochondria and are eventually extruded as potent interferogenic complexes. In support of the in vivo relevance of this phenomenon, mitochondrial retention of Ox nucleoids is a feature of SLE blood neutrophils, and autoantibodies against Ox mtDNA are present in a fraction of patients. This pathway represents a novel therapeutic target in human SLE.
© 2016 Caielli et al.
Figures
Comment in
-
A CD4+ T cell population provides B cell help in systemic lupus erythematosus.Nat Rev Nephrol. 2019 Mar;15(3):123. doi: 10.1038/s41581-018-0096-1. Nat Rev Nephrol. 2019. PMID: 30532021 No abstract available.
References
-
- Bentham J., Morris D.L., Cunninghame Graham D.S., Pinder C.L., Tombleson P., Behrens T.W., Martín J., Fairfax B.P., Knight J.C., Chen L., et al. 2015. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet. 47:1457–1464. 10.1038/ng.3434 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
