

Learning from Co-expression Networks: Possibilities and Challenges
- PMID: 27092161
- PMCID: PMC4825623
- DOI: 10.3389/fpls.2016.00444
Learning from Co-expression Networks: Possibilities and Challenges
Abstract
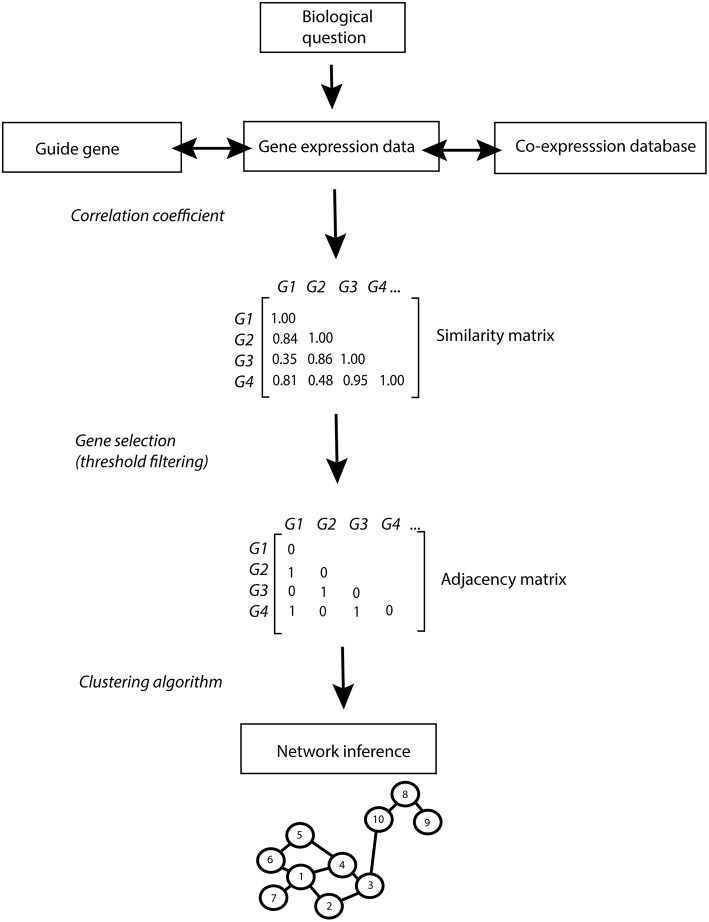
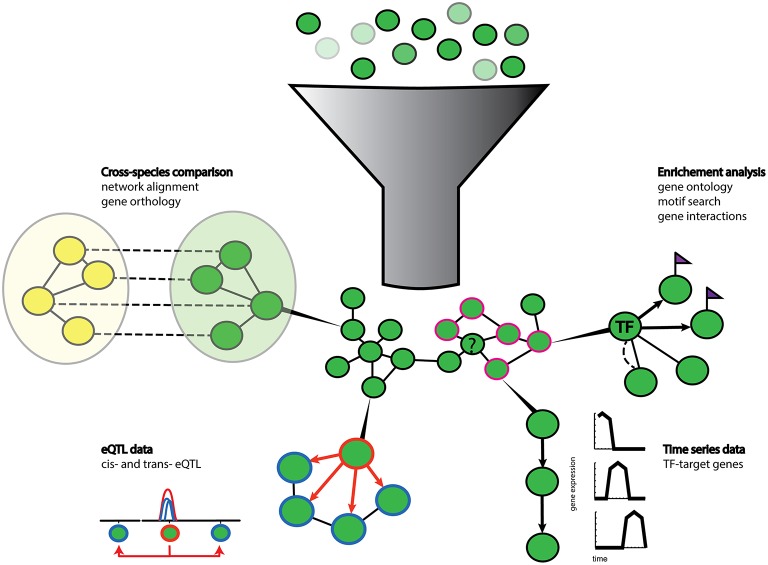
Plants are fascinating and complex organisms. A comprehensive understanding of the organization, function and evolution of plant genes is essential to disentangle important biological processes and to advance crop engineering and breeding strategies. The ultimate aim in deciphering complex biological processes is the discovery of causal genes and regulatory mechanisms controlling these processes. The recent surge of omics data has opened the door to a system-wide understanding of the flow of biological information underlying complex traits. However, dealing with the corresponding large data sets represents a challenging endeavor that calls for the development of powerful bioinformatics methods. A popular approach is the construction and analysis of gene networks. Such networks are often used for genome-wide representation of the complex functional organization of biological systems. Network based on similarity in gene expression are called (gene) co-expression networks. One of the major application of gene co-expression networks is the functional annotation of unknown genes. Constructing co-expression networks is generally straightforward. In contrast, the resulting network of connected genes can become very complex, which limits its biological interpretation. Several strategies can be employed to enhance the interpretation of the networks. A strategy in coherence with the biological question addressed needs to be established to infer reliable networks. Additional benefits can be gained from network-based strategies using prior knowledge and data integration to further enhance the elucidation of gene regulatory relationships. As a result, biological networks provide many more applications beyond the simple visualization of co-expressed genes. In this study we review the different approaches for co-expression network inference in plants. We analyse integrative genomics strategies used in recent studies that successfully identified candidate genes taking advantage of gene co-expression networks. Additionally, we discuss promising bioinformatics approaches that predict networks for specific purposes.
Keywords: co-expression; gene expression; gene networks; gene prioritization; transcriptomics.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials