

Cytochrome P450 1A2 Metabolizes 17β-Estradiol to Suppress Hepatocellular Carcinoma
- PMID: 27093553
- PMCID: PMC4836701
- DOI: 10.1371/journal.pone.0153863
Cytochrome P450 1A2 Metabolizes 17β-Estradiol to Suppress Hepatocellular Carcinoma
Abstract
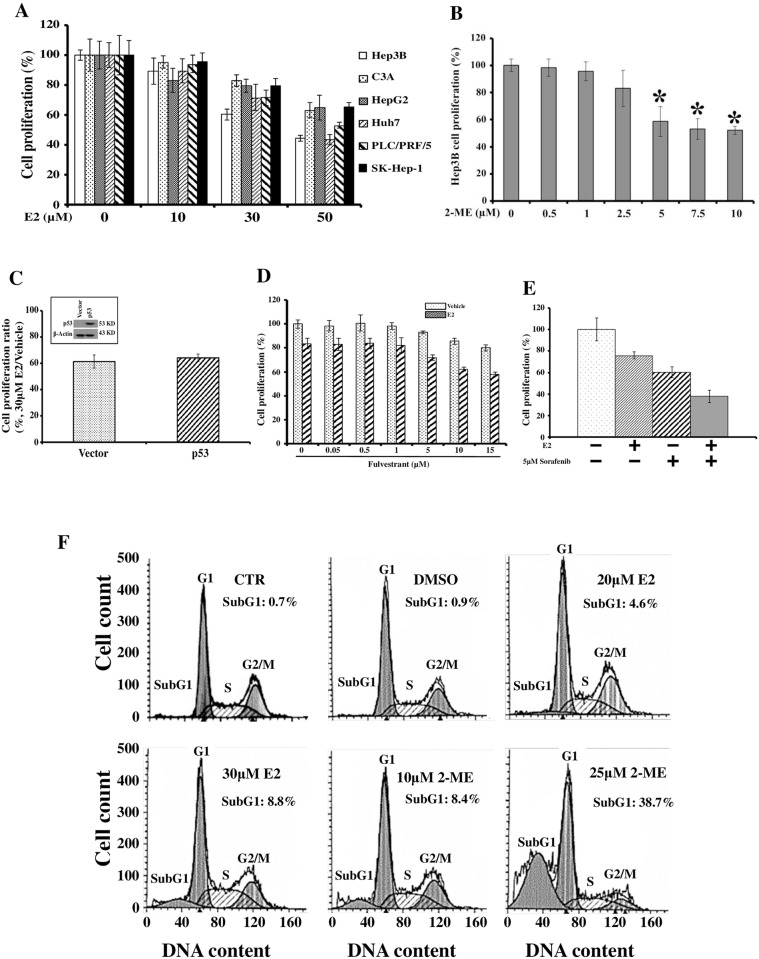
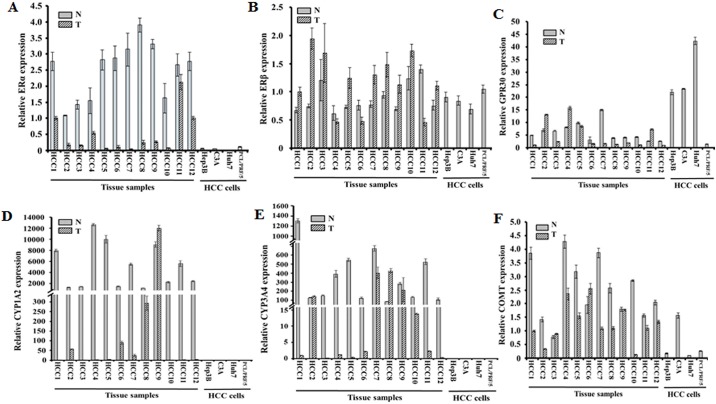
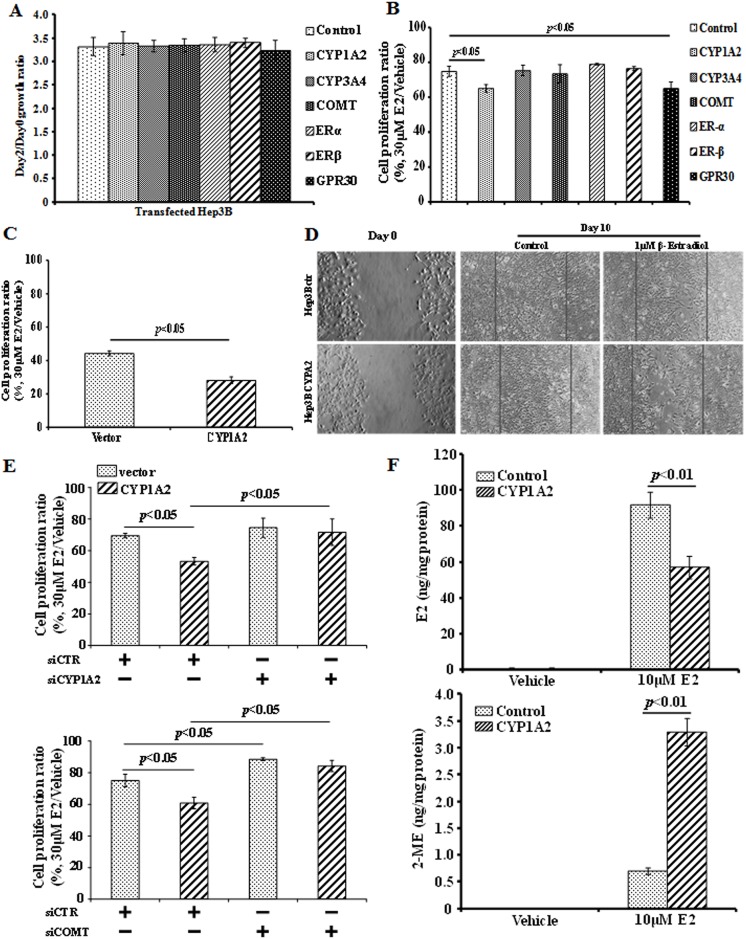
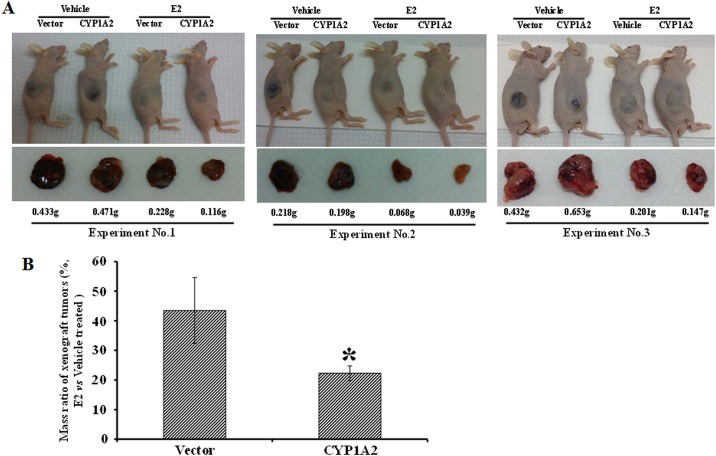
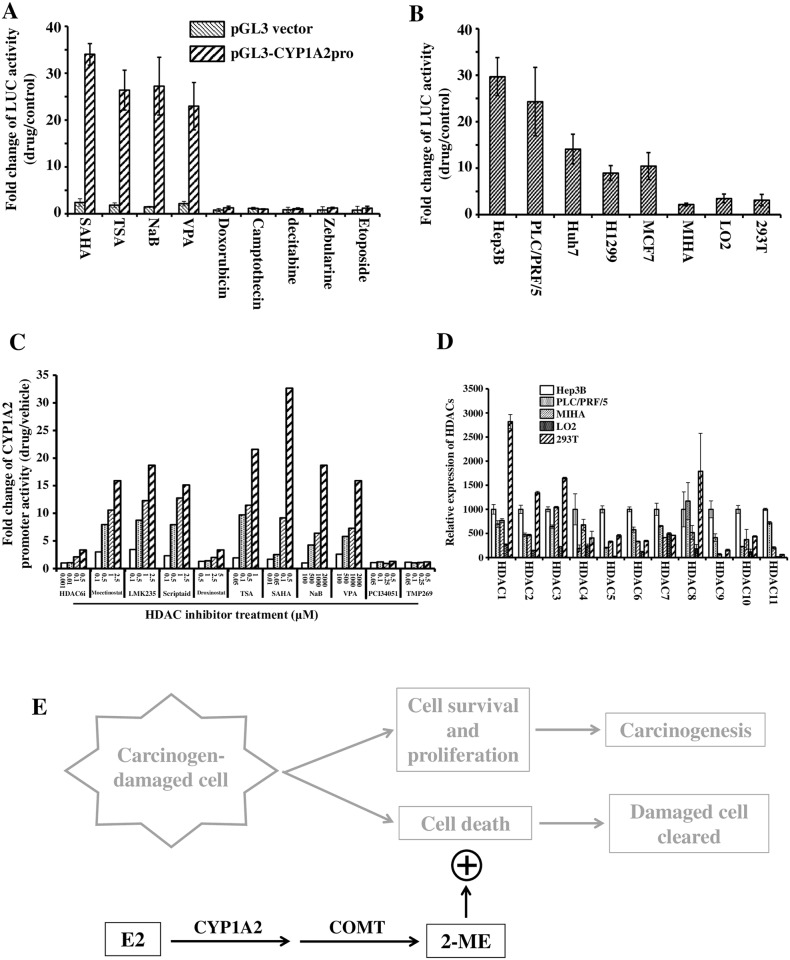
Hepatocellular carcinoma (HCC) occurs more frequently in men than in women. It is commonly agreed that estrogen plays important roles in suppressing HCC development, however, the underlying mechanism remains largely unknown. Since estrogen is mainly metabolized in liver and its metabolites affect cell proliferation, we sought to investigate if the liver-specific cytochrome P450 1A2 (CYP1A2) mediated the inhibitory effect of estrogen on HCC. In this study, the expression of estrogen-metabolizing enzyme CYP1A2 was determined in HCC tissues and cell lines. Cell proliferation and apoptosis were assessed in cells with or without CYP1A2 overexpression. The levels of 17β-estradiol (E2) and its metabolite 2-methoxyestradiol (2-ME) were determined. A xenograft tumor model in mice was established to confirm the findings. It was found that CYP1A2 expression was greatly repressed in HCC. E2 suppressed HCC cell proliferation and xenograft tumor development by inducing apoptosis. The inhibitory effect was significantly enhanced in cells with CYP1A2 overexpression, which effectively conversed E2 to the cytotoxic 2-ME. E2 in combination with sorafenib showed an additive effect on HCC. The anti-HCC effect of E2 was not associated with estrogen receptors ERα and ERβ as well as tumor suppressor P53 but enhanced by the approved anti-HCC drug sorafenib. In addition, HDAC inhibitors greatly induced CYP1A2 promoter activities in cancer cells, especially liver cancer cells, but not in non-tumorigenic cells. Collectively, CYP1A2 metabolizes E2 to generate the potent anti-tumor agent 2-ME in HCC. The reduction of CYP1A2 significantly disrupts this metabolic pathway, contributing the progression and growth of HCC and the gender disparity of this malignancy.
Conflict of interest statement
Figures
References
-
- Park SJ, Jeong SY, Kim HJ. Y chromosome loss and other genomic alterations in hepatocellular carcinoma cell lines analyzed by CGH and CGH array. Cancer Genet Cytogenet. 2006;166: 56–64. - PubMed
-
- Liu J, Wang ZM, Zhen SF, Wu XP, Ma DX, Li ZH, et al. Aberration of X chromosome in liver neoplasm detected by fluorescence in situ hybridization. Hepatobiliary Pancreat Dis Int. 2004;3: 110–114 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
