

Omics Profiling in Precision Oncology
- PMID: 27099341
- PMCID: PMC4974334
- DOI: 10.1074/mcp.O116.059253
Omics Profiling in Precision Oncology
Abstract
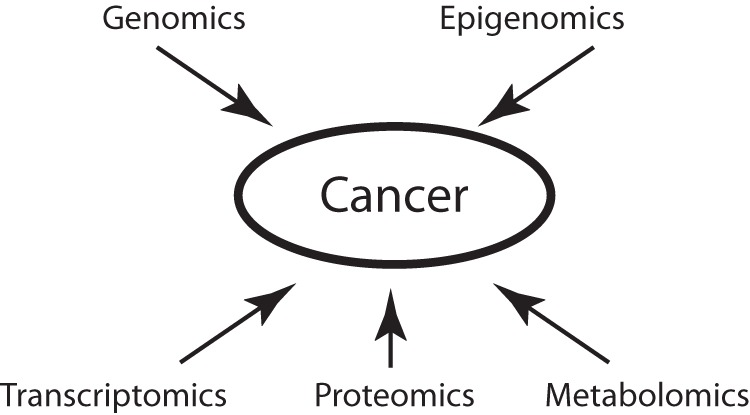
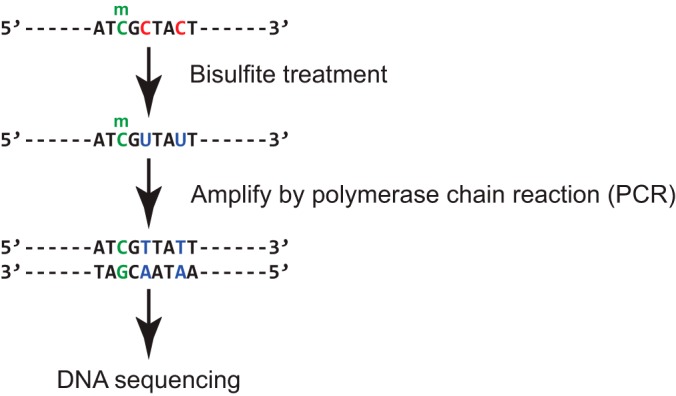
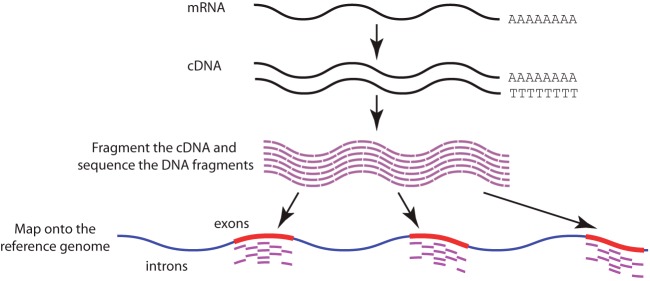
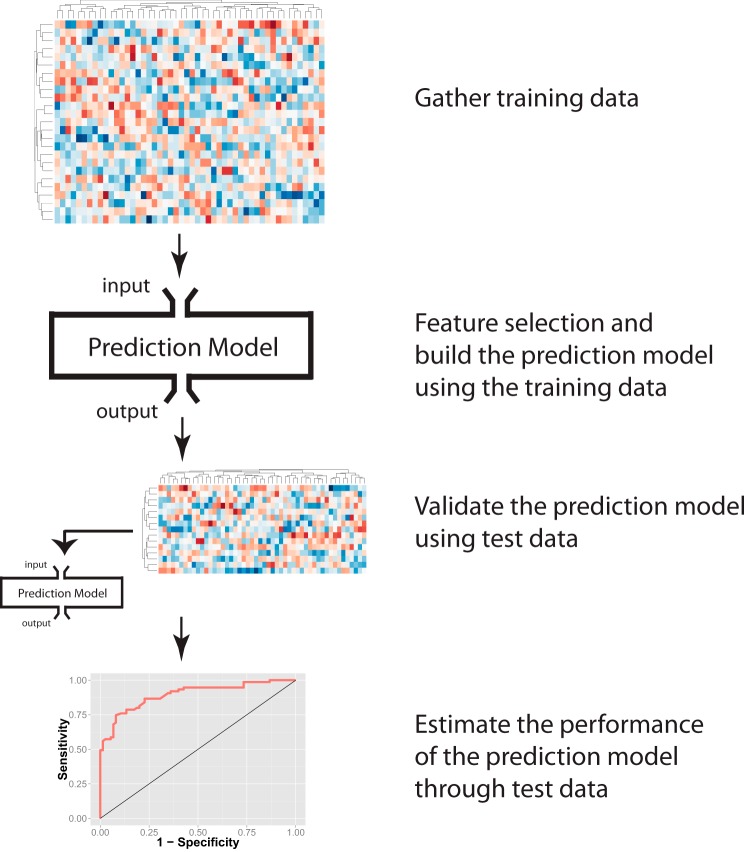
Cancer causes significant morbidity and mortality worldwide, and is the area most targeted in precision medicine. Recent development of high-throughput methods enables detailed omics analysis of the molecular mechanisms underpinning tumor biology. These studies have identified clinically actionable mutations, gene and protein expression patterns associated with prognosis, and provided further insights into the molecular mechanisms indicative of cancer biology and new therapeutics strategies such as immunotherapy. In this review, we summarize the techniques used for tumor omics analysis, recapitulate the key findings in cancer omics studies, and point to areas requiring further research on precision oncology.
© 2016 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
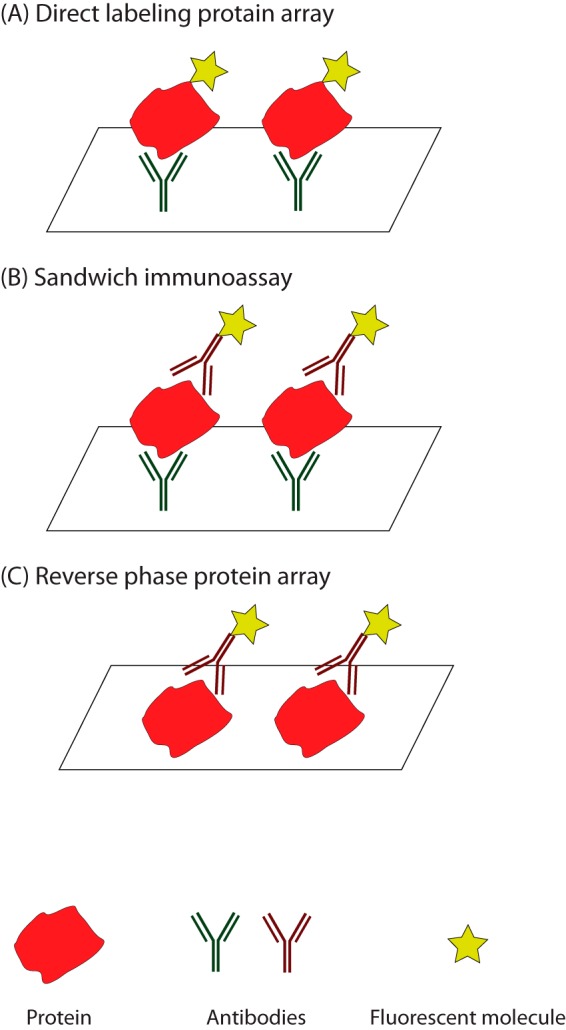
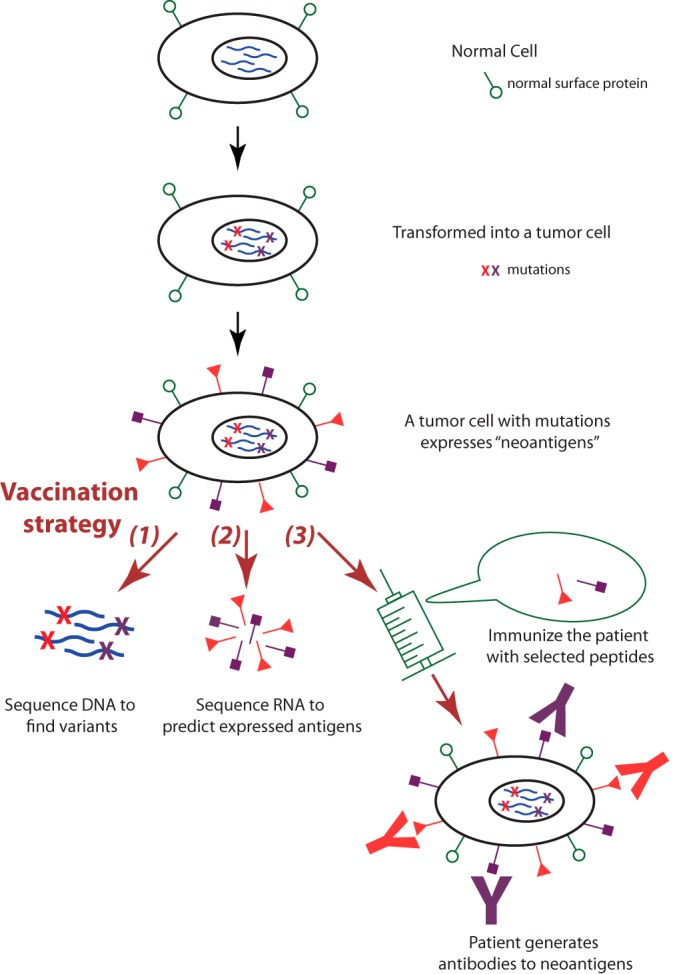
Similar articles
-
The Need for Multi-Omics Biomarker Signatures in Precision Medicine.Int J Mol Sci. 2019 Sep 26;20(19):4781. doi: 10.3390/ijms20194781. Int J Mol Sci. 2019. PMID: 31561483 Free PMC article. Review.
-
A comprehensive review of machine learning techniques for multi-omics data integration: challenges and applications in precision oncology.Brief Funct Genomics. 2024 Sep 27;23(5):549-560. doi: 10.1093/bfgp/elae013. Brief Funct Genomics. 2024. PMID: 38600757 Review.
-
Patient-specific multi-omics models and the application in personalized combination therapy.Future Oncol. 2020 Aug;16(23):1737-1750. doi: 10.2217/fon-2020-0119. Epub 2020 May 28. Future Oncol. 2020. PMID: 32462937 Review.
-
Comprehensive molecular tumor profiling in radiation oncology: How it could be used for precision medicine.Cancer Lett. 2016 Nov 1;382(1):118-126. doi: 10.1016/j.canlet.2016.01.041. Epub 2016 Jan 29. Cancer Lett. 2016. PMID: 26828133 Free PMC article. Review.
-
Multi-omics based artificial intelligence for cancer research.Adv Cancer Res. 2024;163:303-356. doi: 10.1016/bs.acr.2024.06.005. Epub 2024 Jul 9. Adv Cancer Res. 2024. PMID: 39271266 Review.
Cited by
-
Prediction of Target-Drug Therapy by Identifying Gene Mutations in Lung Cancer With Histopathological Stained Image and Deep Learning Techniques.Front Oncol. 2021 Apr 13;11:642945. doi: 10.3389/fonc.2021.642945. eCollection 2021. Front Oncol. 2021. PMID: 33928031 Free PMC article.
-
Predicting Ovarian Cancer Patients' Clinical Response to Platinum-Based Chemotherapy by Their Tumor Proteomic Signatures.J Proteome Res. 2016 Aug 5;15(8):2455-65. doi: 10.1021/acs.jproteome.5b01129. Epub 2016 Jul 8. J Proteome Res. 2016. PMID: 27312948 Free PMC article.
-
Modified SureSelectQXT Target Enrichment Protocol for Illumina Multiplexed Sequencing of FFPE Samples.Biol Proced Online. 2018 Oct 12;20:19. doi: 10.1186/s12575-018-0084-7. eCollection 2018. Biol Proced Online. 2018. PMID: 30337841 Free PMC article.
-
Computational Approaches in Theranostics: Mining and Predicting Cancer Data.Pharmaceutics. 2019 Mar 13;11(3):119. doi: 10.3390/pharmaceutics11030119. Pharmaceutics. 2019. PMID: 30871264 Free PMC article. Review.
-
Quantitative SWATH-Based Proteomic Profiling for Identification of Mechanism-Driven Diagnostic Biomarkers Conferring in the Progression of Metastatic Prostate Cancer.Front Oncol. 2020 Apr 8;10:493. doi: 10.3389/fonc.2020.00493. eCollection 2020. Front Oncol. 2020. PMID: 32322560 Free PMC article.
References
-
- Torre L. A., Bray F., Siegel R. L., Ferlay J., Lortet-Tieulent J., and Jemal A. (2015) Global cancer statistics, 2012. CA 65, 87–108 - PubMed
-
- Siegel R. L., Miller K. D., and Jemal A. (2015) Cancer statistics, 2015. CA 65, 5–29 - PubMed
-
- Hanahan D., and Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 - PubMed
-
- Hanahan D., and Weinberg R. A. (2000) The hallmarks of cancer. Cell 100, 57–70 - PubMed
-
- National Research Council Committee on A Framework for Developing a New Taxonomy of Disease (2011) Toward precision medicine: Building a knowledge network for biomedical research and a new taxonomy of disease, National Academies Press (US) - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
