

Quantitative Tagless Copurification: A Method to Validate and Identify Protein-Protein Interactions
- PMID: 27099342
- PMCID: PMC5083090
- DOI: 10.1074/mcp.M115.057117
Quantitative Tagless Copurification: A Method to Validate and Identify Protein-Protein Interactions
Abstract
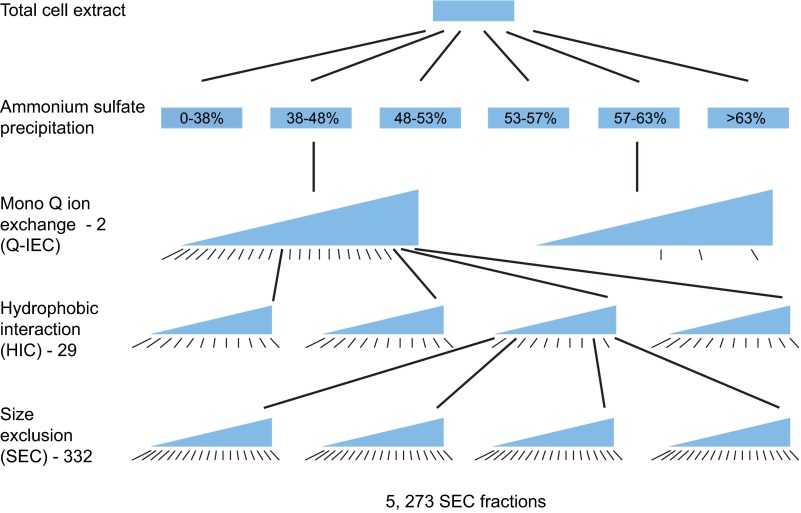
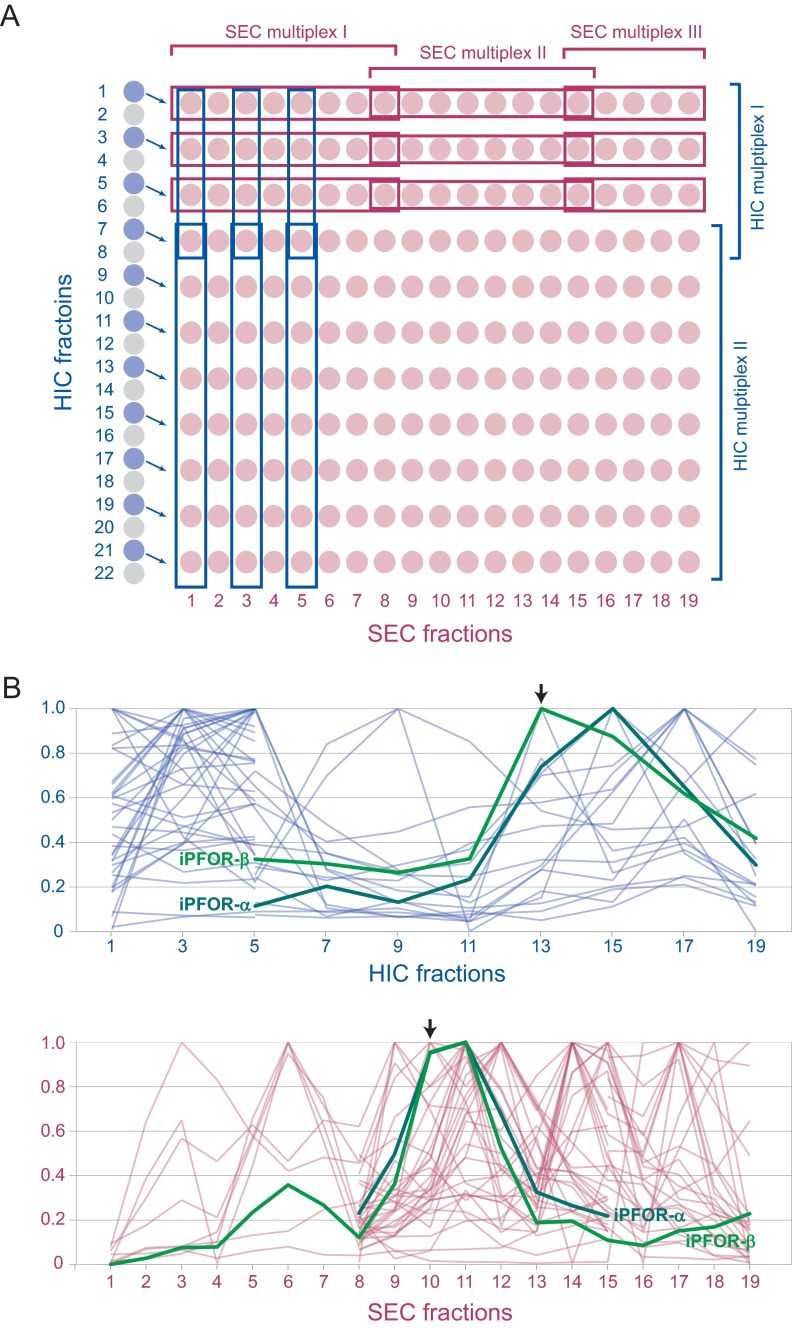
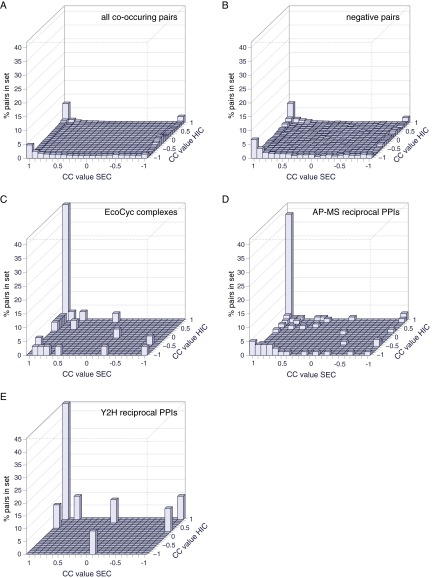
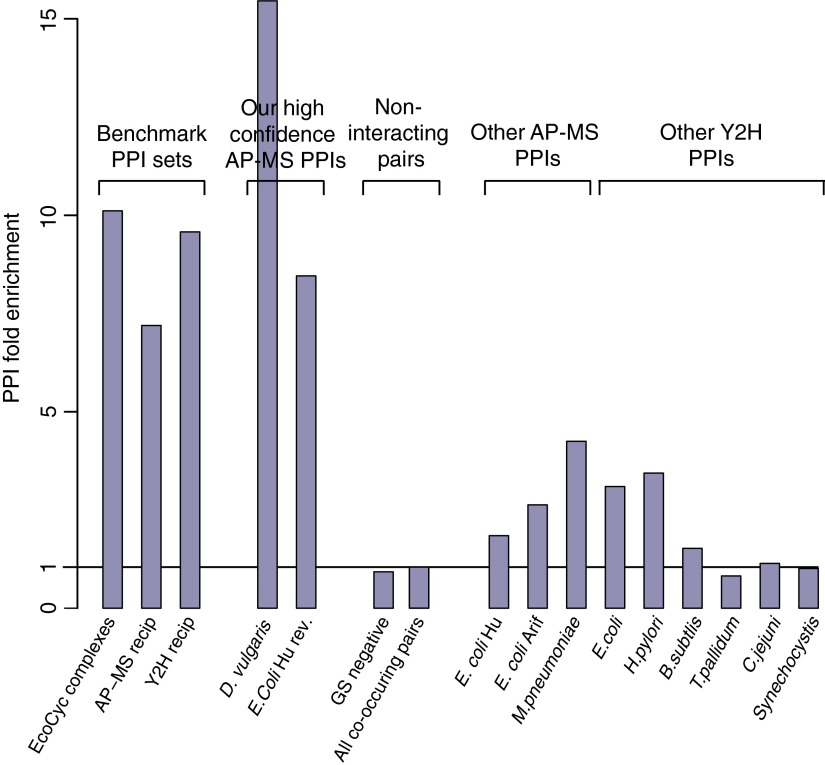
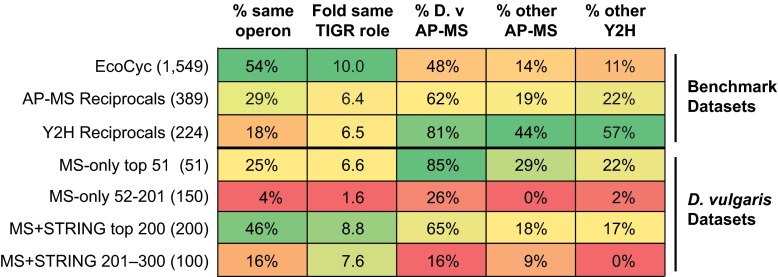
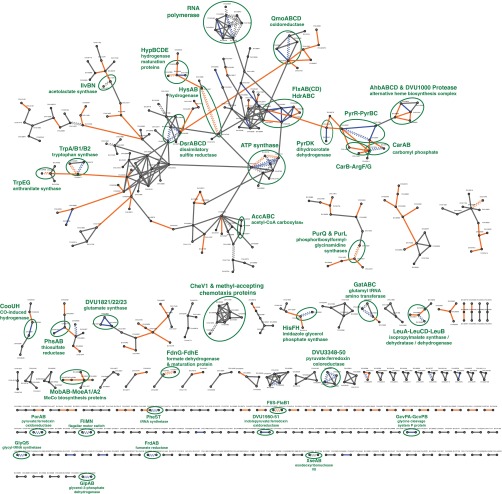
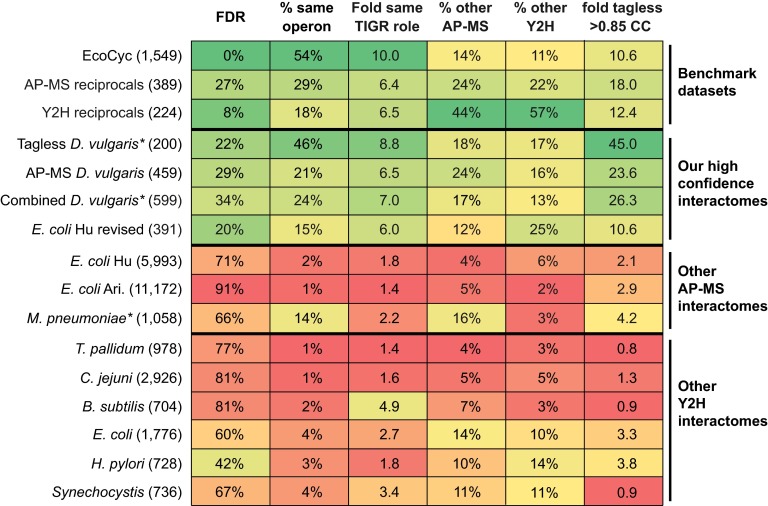
Identifying protein-protein interactions (PPIs) at an acceptable false discovery rate (FDR) is challenging. Previously we identified several hundred PPIs from affinity purification - mass spectrometry (AP-MS) data for the bacteria Escherichia coli and Desulfovibrio vulgaris These two interactomes have lower FDRs than any of the nine interactomes proposed previously for bacteria and are more enriched in PPIs validated by other data than the nine earlier interactomes. To more thoroughly determine the accuracy of ours or other interactomes and to discover further PPIs de novo, here we present a quantitative tagless method that employs iTRAQ MS to measure the copurification of endogenous proteins through orthogonal chromatography steps. 5273 fractions from a four-step fractionation of a D. vulgaris protein extract were assayed, resulting in the detection of 1242 proteins. Protein partners from our D. vulgaris and E. coli AP-MS interactomes copurify as frequently as pairs belonging to three benchmark data sets of well-characterized PPIs. In contrast, the protein pairs from the nine other bacterial interactomes copurify two- to 20-fold less often. We also identify 200 high confidence D. vulgaris PPIs based on tagless copurification and colocalization in the genome. These PPIs are as strongly validated by other data as our AP-MS interactomes and overlap with our AP-MS interactome for D.vulgaris within 3% of expectation, once FDRs and false negative rates are taken into account. Finally, we reanalyzed data from two quantitative tagless screens of human cell extracts. We estimate that the novel PPIs reported in these studies have an FDR of at least 85% and find that less than 7% of the novel PPIs identified in each screen overlap. Our results establish that a quantitative tagless method can be used to validate and identify PPIs, but that such data must be analyzed carefully to minimize the FDR.
© 2016 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Alberts B., Johnson A., Lewis J., Raff M., Roberts K., and Walter P. (2007) Molecular Biol. Cell, 5 edition ed., Garland Science, New York.
-
- Kristensen A. R., and Foster L. J. (2013) High throughput strategies for probing the different organizational levels of protein interaction networks. Mol. bioSystems 9, 2201–2212 - PubMed
-
- von Mering C., Krause R., Snel B., Cornell M., Oliver S. G., Fields S., and Bork P. (2002) Comparative assessment of large-scale data sets of protein-protein interactions. Nature 417, 399–403 - PubMed
-
- Edwards A. M., Kus B., Jansen R., Greenbaum D., Greenblatt J., and Gerstein M. (2002) Bridging structural biology and genomics: assessing protein interaction data with known complexes. Trends Gen. 18, 529–536 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
