

Clonify: unseeded antibody lineage assignment from next-generation sequencing data
- PMID: 27102563
- PMCID: PMC4840318
- DOI: 10.1038/srep23901
Clonify: unseeded antibody lineage assignment from next-generation sequencing data
Abstract
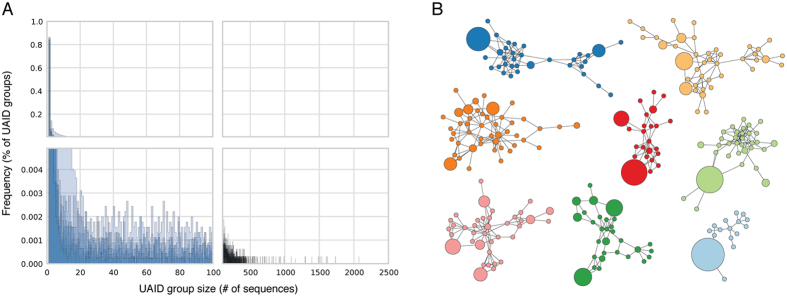
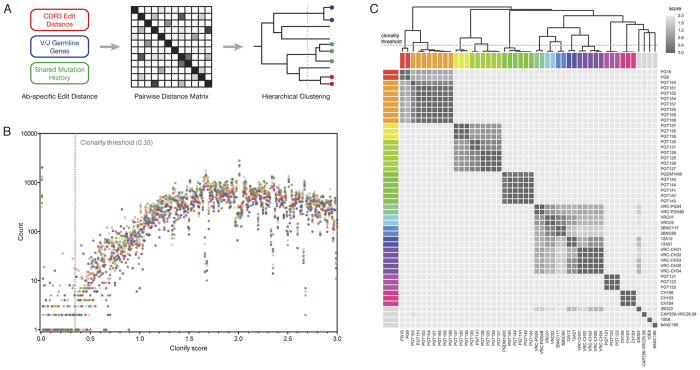
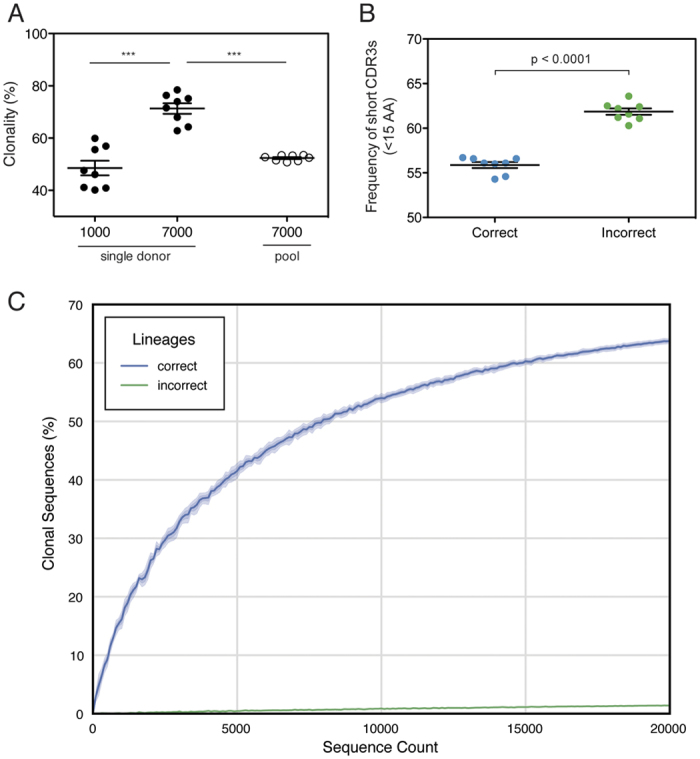
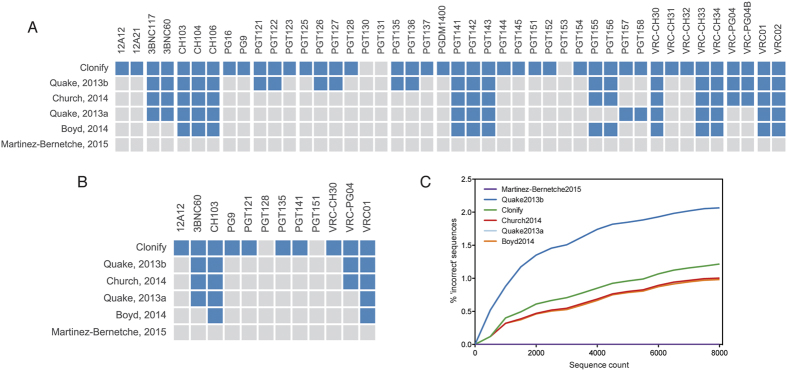
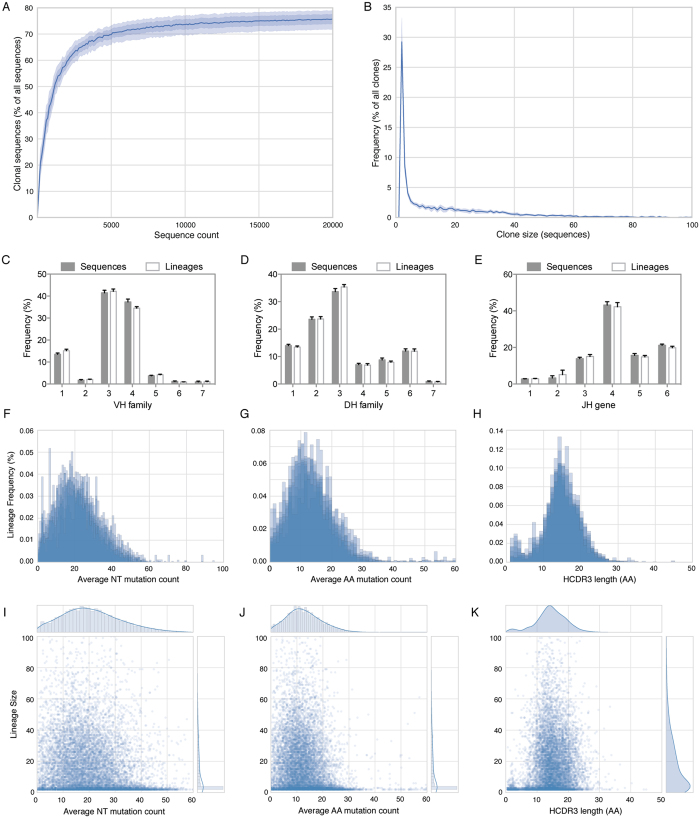
Defining the dynamics and maturation processes of antibody clonal lineages is crucial to understanding the humoral response to infection and immunization. Although individual antibody lineages have been previously analyzed in isolation, these studies provide only a narrow view of the total antibody response. Comprehensive study of antibody lineages has been limited by the lack of an accurate clonal lineage assignment algorithm capable of operating on next-generation sequencing datasets. To address this shortcoming, we developed Clonify, which is able to perform unseeded lineage assignment on very large sets of antibody sequences. Application of Clonify to IgG+ memory repertoires from healthy individuals revealed a surprising lack of influence of large extended lineages on the overall repertoire composition, indicating that this composition is driven less by the order and frequency of pathogen encounters than previously thought. Clonify is freely available at www.github.com/briney/clonify-python.
Figures
References
-
- Koff W. C., Gust I. D. & Plotkin S. A. Toward a human vaccines project. Nat Immunol 15, 589–592 (2014). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
