

Context-dependent effects of SOCS3 in angiotensin II-induced vascular dysfunction and hypertension in mice: mechanisms and role of bone marrow-derived cells
- PMID: 27106041
- PMCID: PMC4967211
- DOI: 10.1152/ajpheart.00204.2016
Context-dependent effects of SOCS3 in angiotensin II-induced vascular dysfunction and hypertension in mice: mechanisms and role of bone marrow-derived cells
Abstract
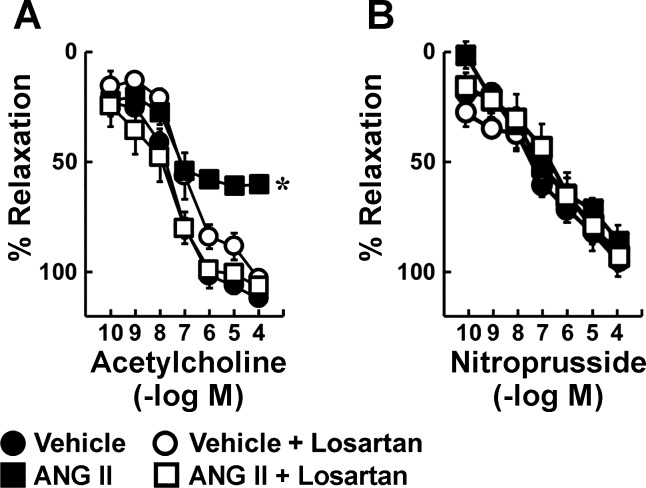
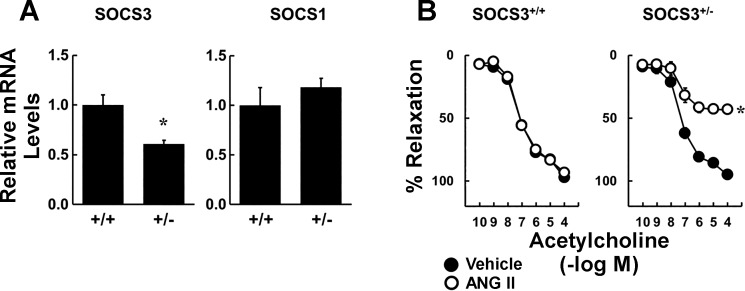
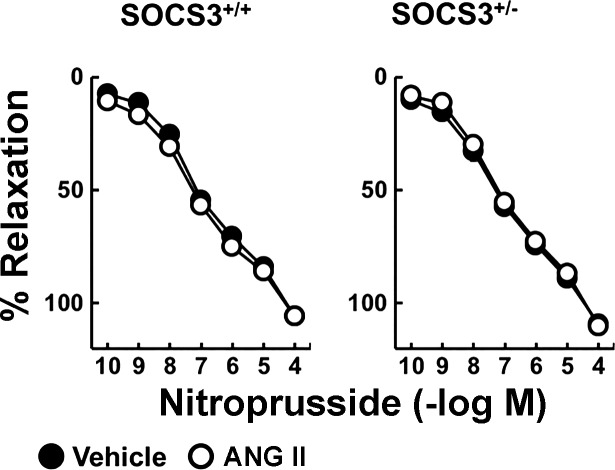
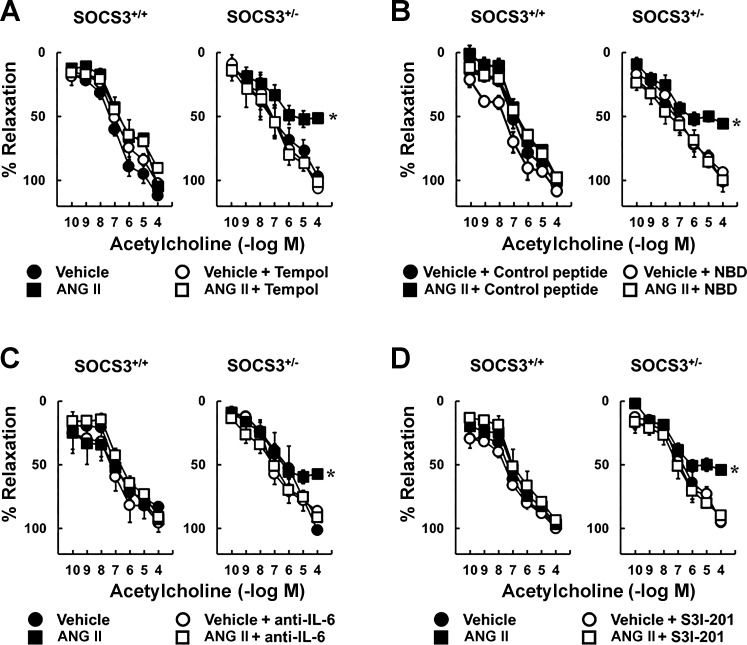
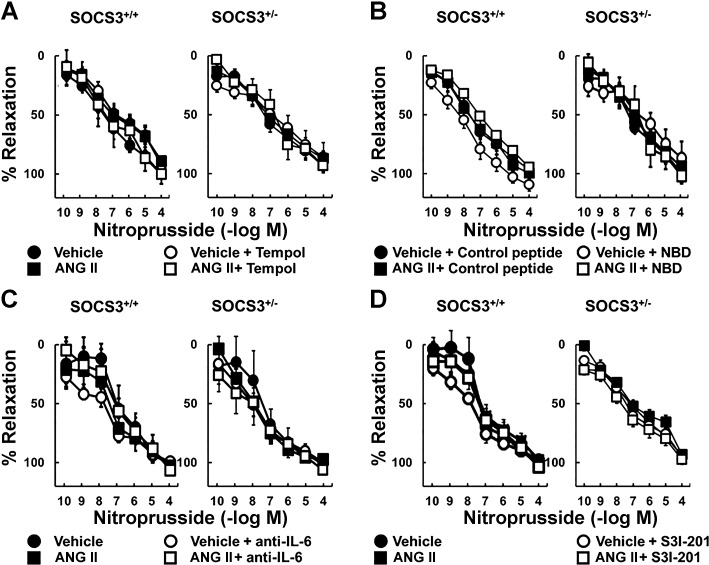
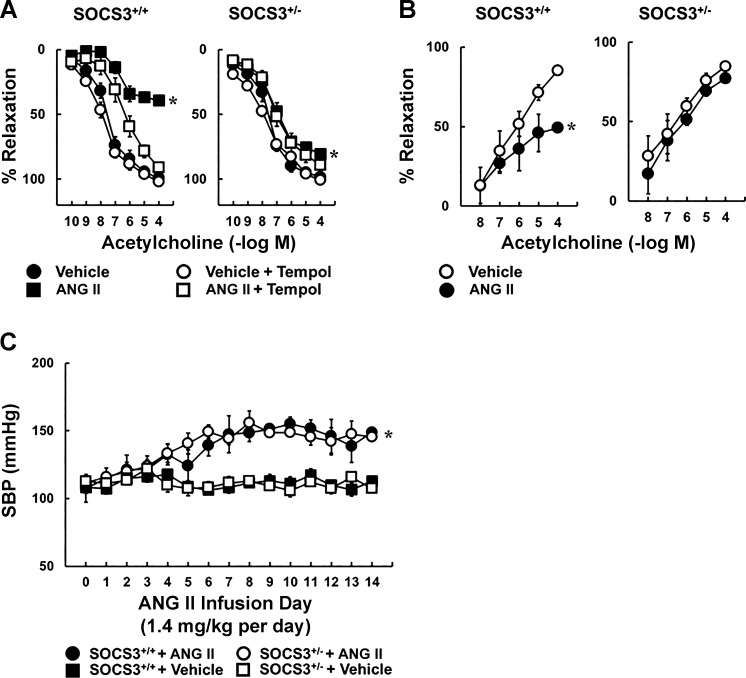
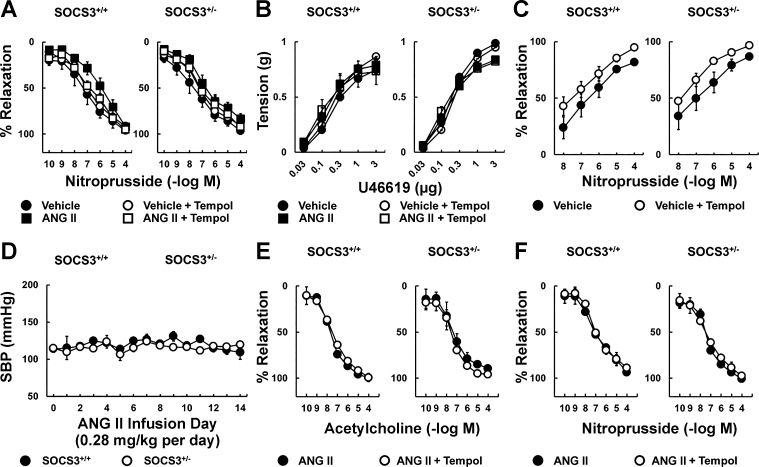
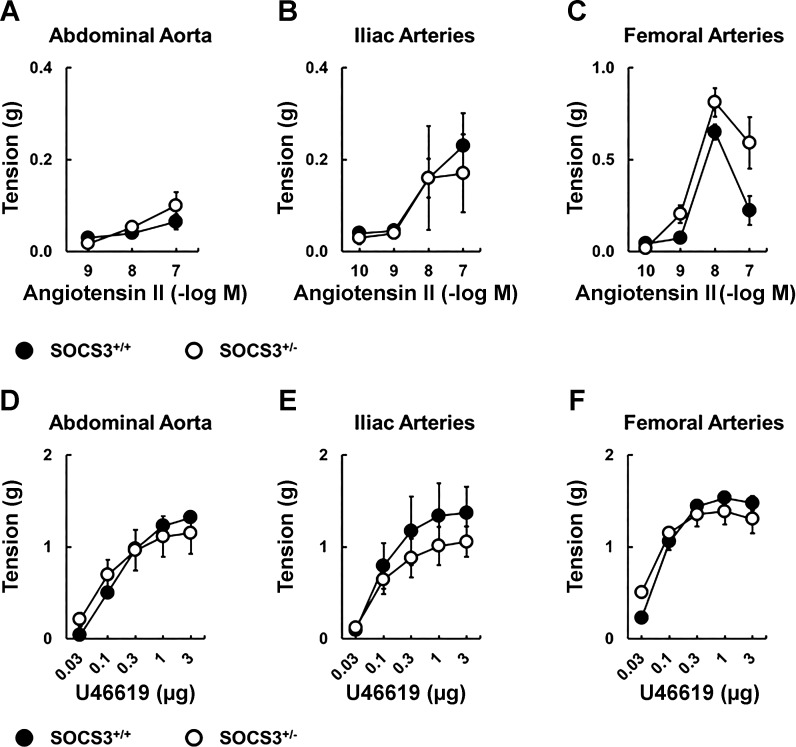
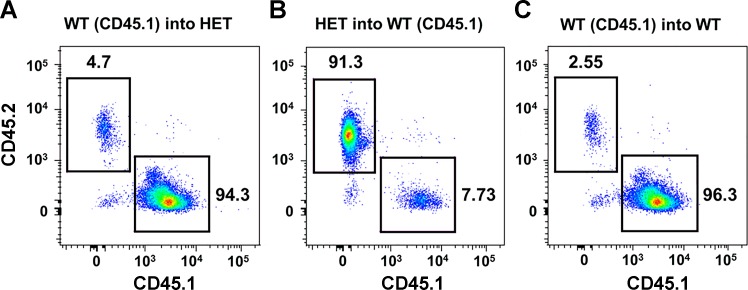
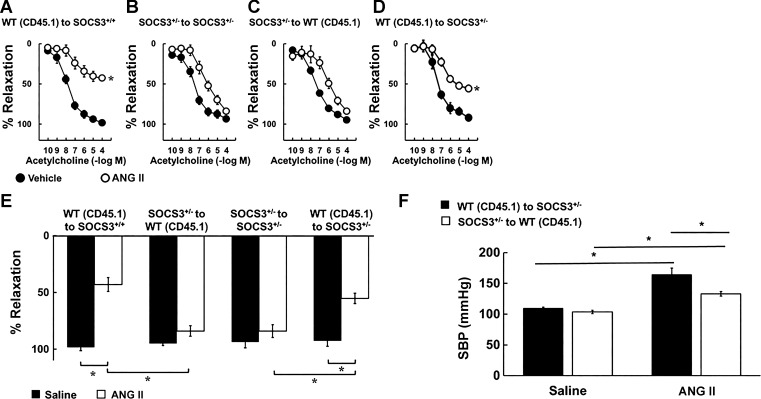
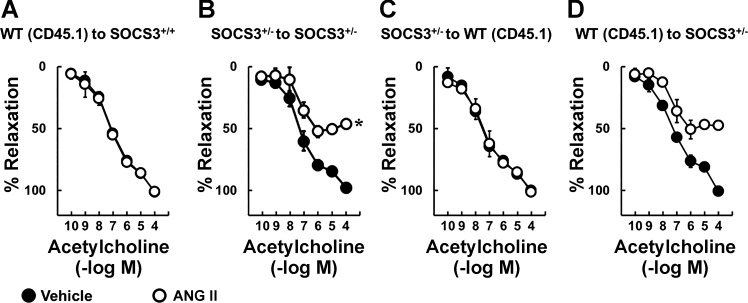
Carotid artery disease is a major contributor to stroke and cognitive deficits. Angiotensin II (Ang II) promotes vascular dysfunction and disease through mechanisms that include the IL-6/STAT3 pathway. Here, we investigated the importance of suppressor of cytokine signaling 3 (SOCS3) in models of Ang II-induced vascular dysfunction. We examined direct effects of Ang II on carotid arteries from SOCS3-deficient (SOCS3(+/-)) mice and wild-type (WT) littermates using organ culture and then tested endothelial function with acetylcholine (ACh). A low concentration of Ang II (1 nmol/l) did not affect ACh-induced vasodilation in WT but reduced that of SOCS3(+/-) mice by ∼50% (P < 0.05). In relation to mechanisms, effects of Ang II in SOCS3(+/-) mice were prevented by inhibitors of STAT3, IL-6, NF-κB, or superoxide. Systemic Ang II (1.4 mg/kg per day for 14 days) also reduced vasodilation to ACh in WT. Surprisingly, SOCS3 deficiency prevented most of the endothelial dysfunction. To examine potential underlying mechanisms, we performed bone marrow transplantation. WT mice reconstituted with SOCS3(+/-) bone marrow were protected from Ang II-induced endothelial dysfunction, whereas reconstitution of SOCS3(+/-) mice with WT bone marrow exacerbated Ang II-induced effects. The SOCS3 genotype of bone marrow-derived cells did not influence direct effects of Ang II on vascular function. These data provide new mechanistic insight into the influence of SOCS3 on the vasculature, including divergent effects depending on the source of Ang II. Bone marrow-derived cells deficient in SOCS3 protect against systemic Ang II-induced vascular dysfunction.
Keywords: carotid artery disease; cerebral arteries; endothelial dysfunction; endothelium; suppressor of cytokine signaling 3.
Copyright © 2016 the American Physiological Society.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous