

p58(IPK) suppresses NLRP3 inflammasome activation and IL-1β production via inhibition of PKR in macrophages
- PMID: 27113095
- PMCID: PMC4845006
- DOI: 10.1038/srep25013
p58(IPK) suppresses NLRP3 inflammasome activation and IL-1β production via inhibition of PKR in macrophages
Abstract
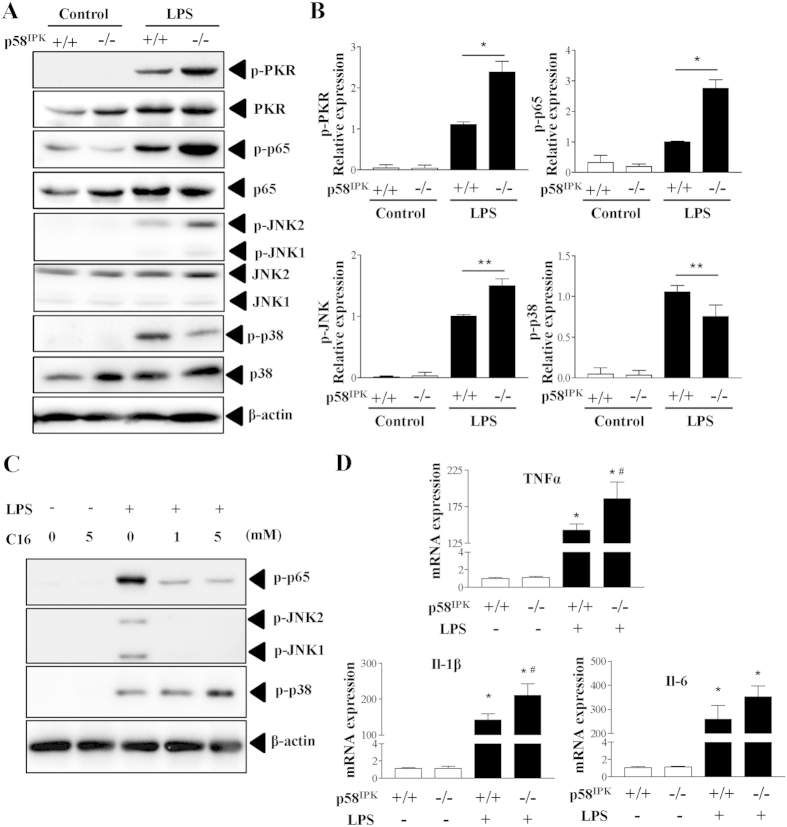
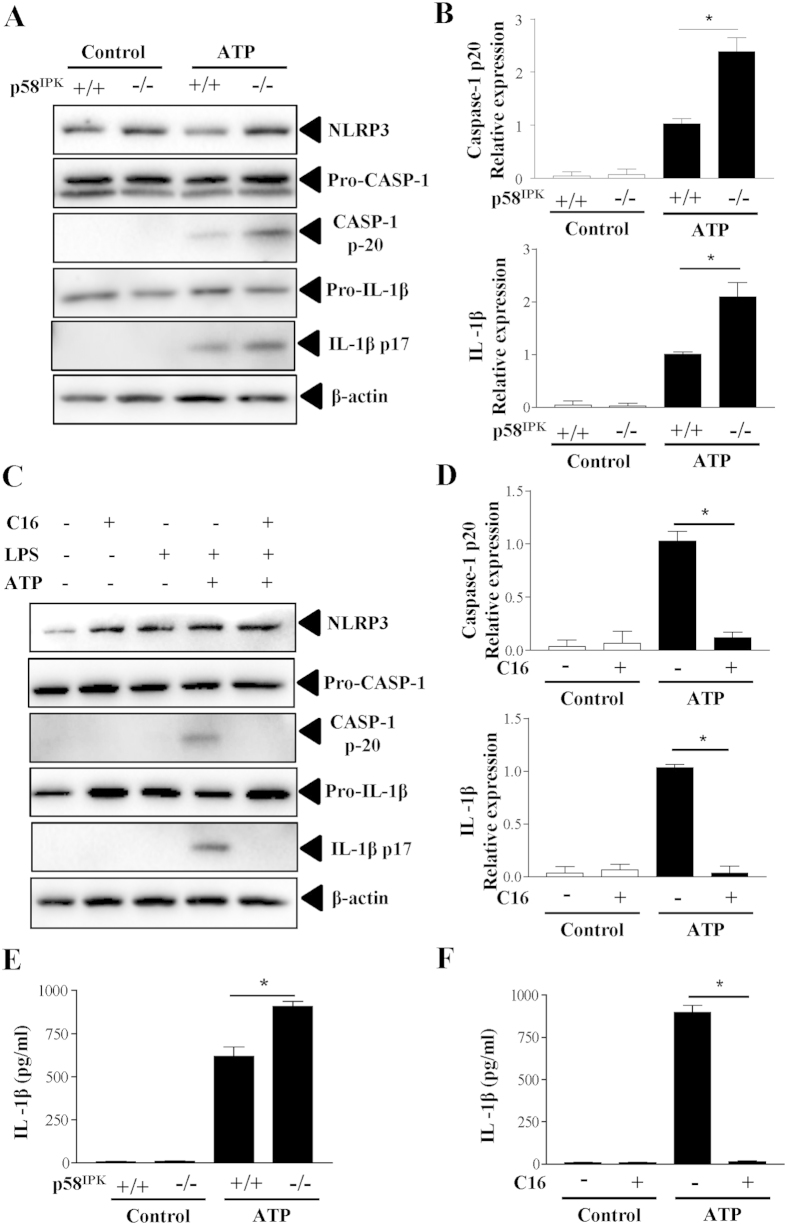
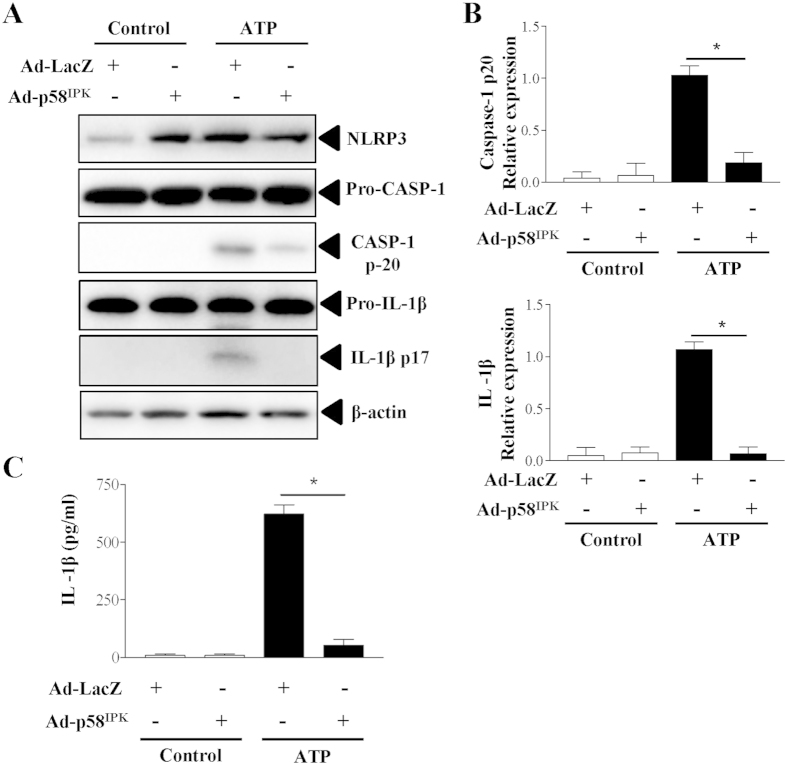
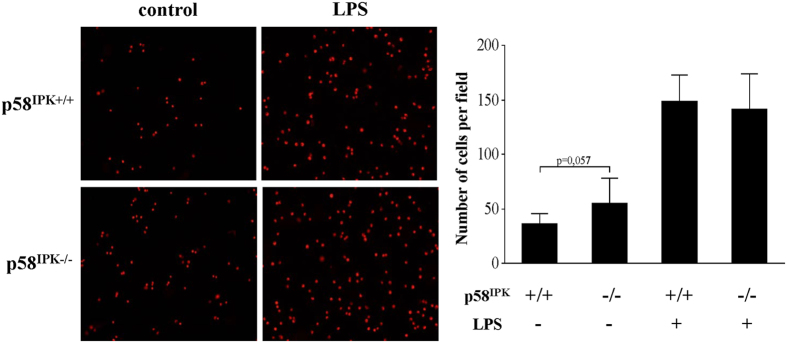
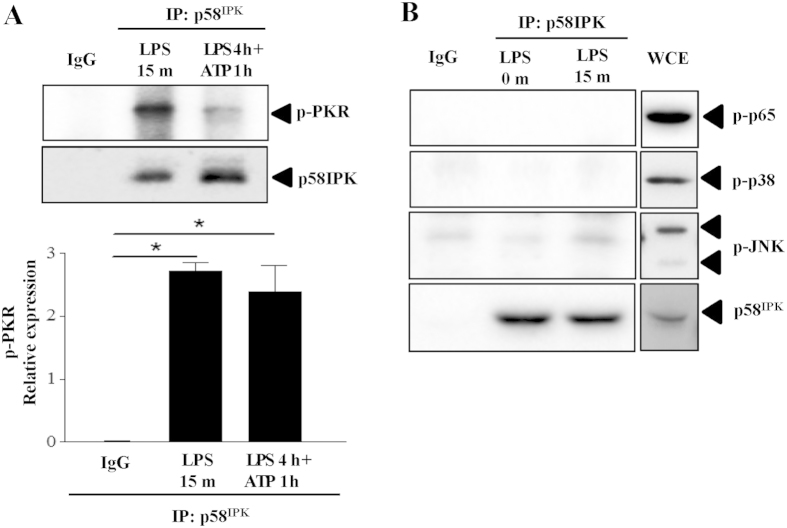
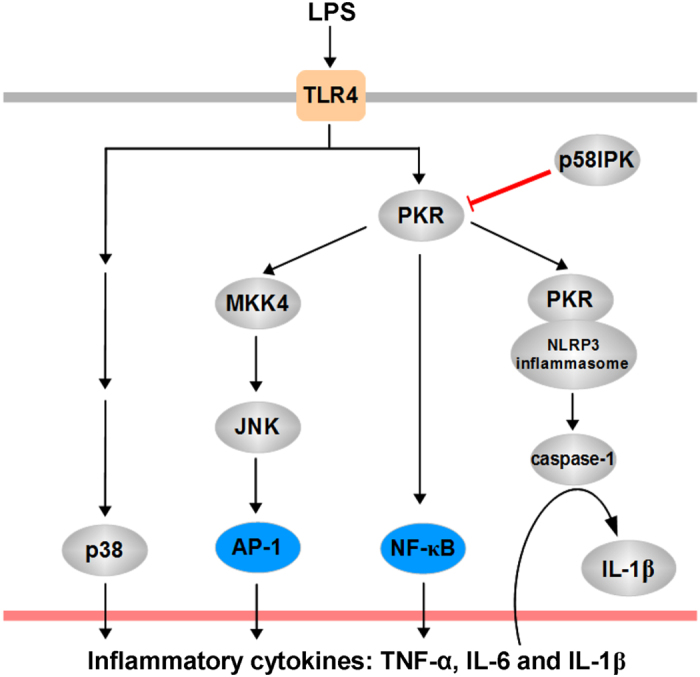
The NLRP3 inflammasome activation is a key signaling event for activation and secretion of pro-inflammatory cytokines such as IL-1β from macrophages. p58(IPK) is a molecular chaperone that regulates protein homeostasis through inhibiting eIF-2α kinases including double-stranded RNA-dependent protein kinase (PKR), which has been recently implicated in inflammasome activation. Herein we investigate the role of p58(IPK) in TLR4 signaling and inflammasome activation in macrophages. Primary bone marrow-derived macrophages (BMDM) was isolated from p58(IPK) knockout (KO) and wildtype (WT) mice and treated with lipopolysaccharide (LPS) and ATP to activate TLR4 signaling and stimulate inflammasome activation. Compared to WT macrophages, p58(IPK) deficient cells demonstrated significantly stronger activation of PKR, NF-κB, and JNK and higher expression of pro-inflammatory genes TNF-α and IL-1β. Coincidently, p58(IPK) deletion intensified NLRP3-inflammasome activation indicated by enhanced caspase 1 cleavage and increased IL-1β maturation and secretion. Pretreatment with specific PKR inhibitor or overexpression of p58(IPK) largely abolished the changes in inflammasome activation and IL-1β secretion in p58(IPK) null macrophages. Furthermore, immunoprecipitation assay confirmed the binding of p58(IPK) with PKR, but not other TLR4 downstream signaling molecules. Collectively, these results suggest a novel and crucial role of p58(IPK) in regulation of inflammasome activation and IL-1β secretion in macrophages.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
