

Tryptophan-2,3-dioxygenase (TDO) inhibition ameliorates neurodegeneration by modulation of kynurenine pathway metabolites
- PMID: 27114543
- PMCID: PMC4868470
- DOI: 10.1073/pnas.1604453113
Tryptophan-2,3-dioxygenase (TDO) inhibition ameliorates neurodegeneration by modulation of kynurenine pathway metabolites
Abstract
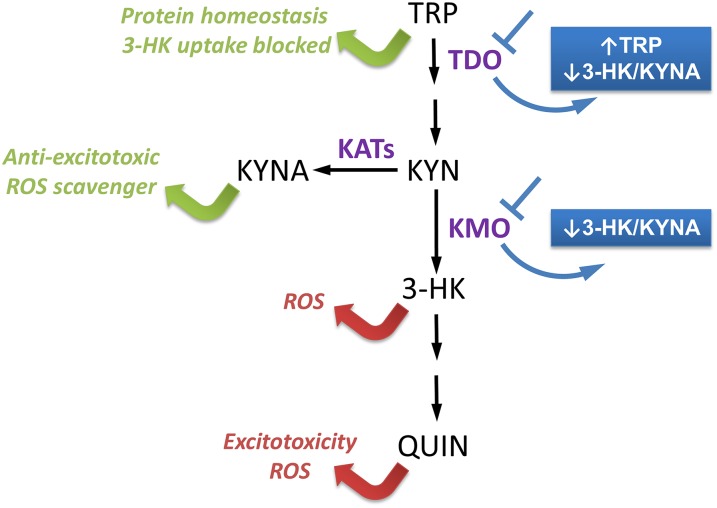
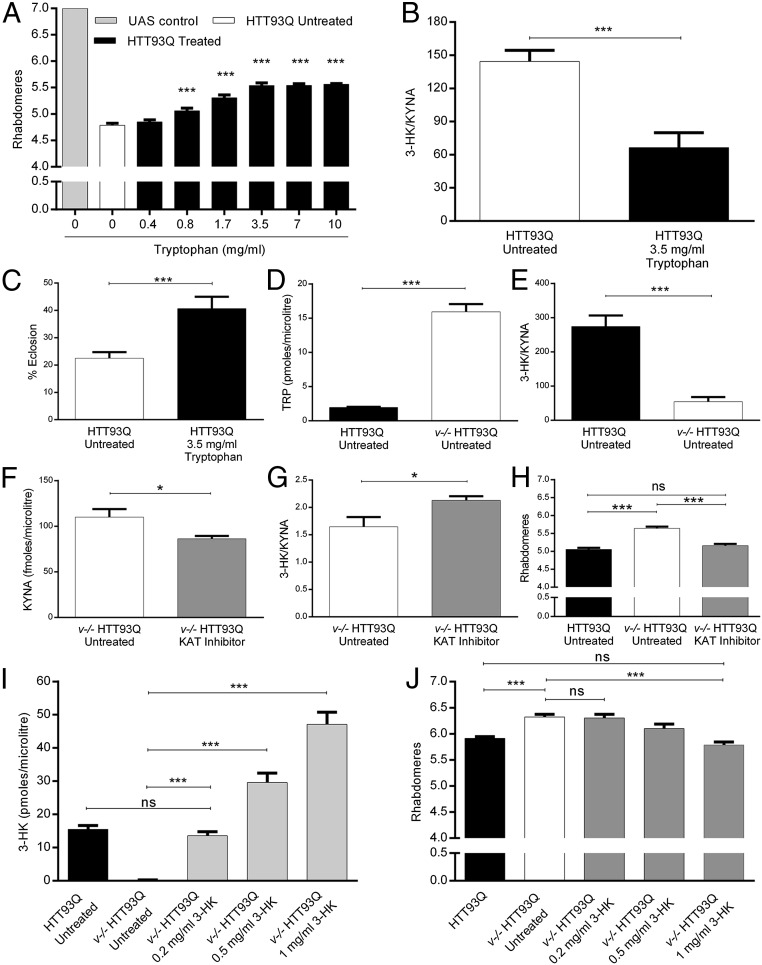
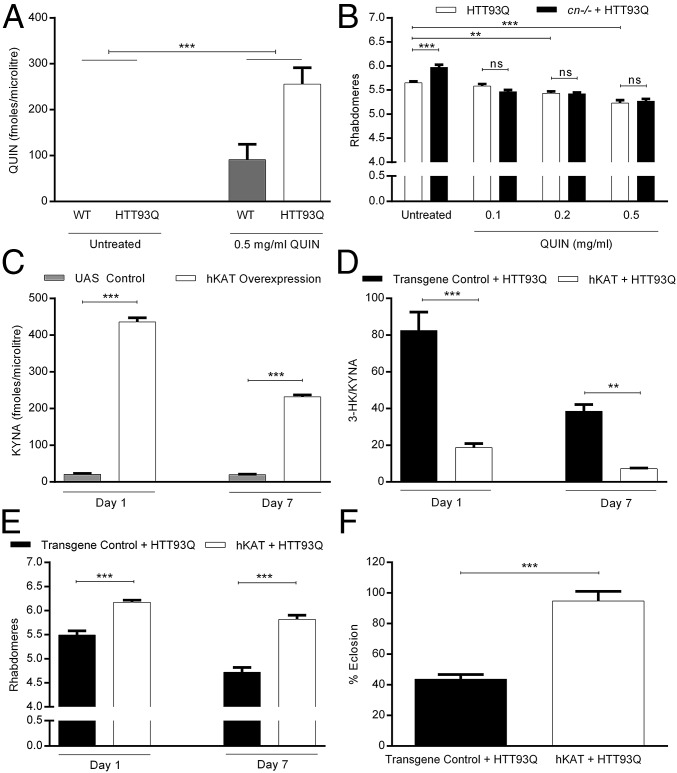
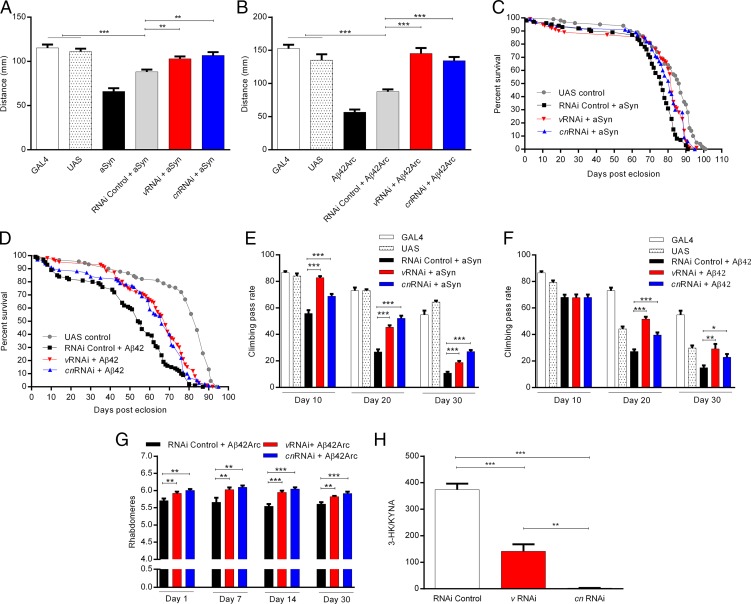
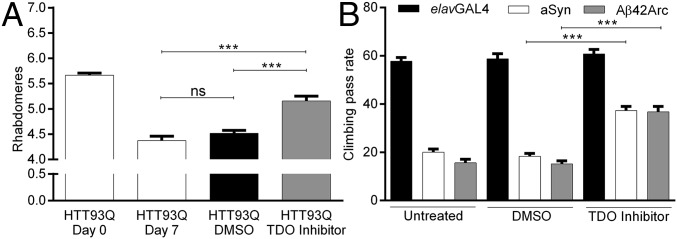
Metabolites of the kynurenine pathway (KP) of tryptophan (TRP) degradation have been closely linked to the pathogenesis of several neurodegenerative disorders. Recent work has highlighted the therapeutic potential of inhibiting two critical regulatory enzymes in this pathway-kynurenine-3-monooxygenase (KMO) and tryptophan-2,3-dioxygenase (TDO). Much evidence indicates that the efficacy of KMO inhibition arises from normalizing an imbalance between neurotoxic [3-hydroxykynurenine (3-HK); quinolinic acid (QUIN)] and neuroprotective [kynurenic acid (KYNA)] KP metabolites. However, it is not clear if TDO inhibition is protective via a similar mechanism or if this is instead due to increased levels of TRP-the substrate of TDO. Here, we find that increased levels of KYNA relative to 3-HK are likely central to the protection conferred by TDO inhibition in a fruit fly model of Huntington's disease and that TRP treatment strongly reduces neurodegeneration by shifting KP flux toward KYNA synthesis. In fly models of Alzheimer's and Parkinson's disease, we provide genetic evidence that inhibition of TDO or KMO improves locomotor performance and ameliorates shortened life span, as well as reducing neurodegeneration in Alzheimer's model flies. Critically, we find that treatment with a chemical TDO inhibitor is robustly protective in these models. Consequently, our work strongly supports targeting of the KP as a potential treatment strategy for several major neurodegenerative disorders and suggests that alterations in the levels of neuroactive KP metabolites could underlie several therapeutic benefits.
Keywords: Alzheimer’s disease; KMO; Parkinson’s disease; TDO; neurodegeneration.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Amaral M, Outeiro TF, Scrutton NS, Giorgini F. The causative role and therapeutic potential of the kynurenine pathway in neurodegenerative disease. J Mol Med (Berl) 2013;91(6):705–713. - PubMed
-
- Thevandavakkam MA, Schwarcz R, Muchowski PJ, Giorgini F. Targeting kynurenine 3-monooxygenase (KMO): Implications for therapy in Huntington’s disease. CNS Neurol Disord Drug Targets. 2010;9(6):791–800. - PubMed
-
- Stone TW, Perkins MN. Quinolinic acid: A potent endogenous excitant at amino acid receptors in CNS. Eur J Pharmacol. 1981;72(4):411–412. - PubMed
-
- Schwarcz R, Whetsell WO, Jr, Mangano RM. Quinolinic acid: An endogenous metabolite that produces axon-sparing lesions in rat brain. Science. 1983;219(4582):316–318. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
