

A Selective Transforming Growth Factor-β Ligand Trap Attenuates Pulmonary Hypertension
- PMID: 27115515
- PMCID: PMC5114445
- DOI: 10.1164/rccm.201510-1955OC
A Selective Transforming Growth Factor-β Ligand Trap Attenuates Pulmonary Hypertension
Abstract
Rationale: Transforming growth factor-β (TGF-β) ligands signal via type I and type II serine-threonine kinase receptors to regulate broad transcriptional programs. Excessive TGF-β-mediated signaling is implicated in the pathogenesis of pulmonary arterial hypertension, based in part on the ability of broad inhibition of activin-like kinase (ALK) receptors 4/5/7 recognizing TGF-β, activin, growth and differentiation factor, and nodal ligands to attenuate experimental pulmonary hypertension (PH). These broad inhibition strategies do not delineate the specific contribution of TGF-β versus a multitude of other ligands, and their translation is limited by cardiovascular and systemic toxicity.
Objectives: We tested the impact of a soluble TGF-β type II receptor extracellular domain expressed as an immunoglobulin-Fc fusion protein (TGFBRII-Fc), serving as a selective TGF-β1/3 ligand trap, in several experimental PH models.
Methods: Signaling studies used cultured human pulmonary artery smooth muscle cells. PH was studied in monocrotaline-treated Sprague-Dawley rats, SU5416/hypoxia-treated Sprague-Dawley rats, and SU5416/hypoxia-treated C57BL/6 mice. PH, cardiac function, vascular remodeling, and valve structure were assessed by ultrasound, invasive hemodynamic measurements, and histomorphometry.
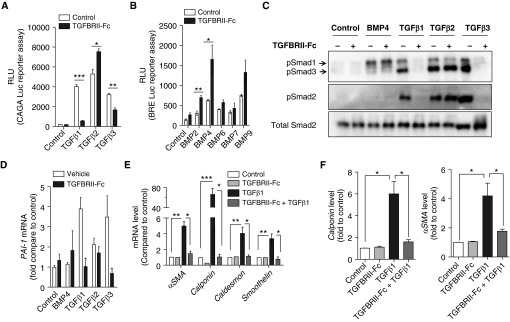
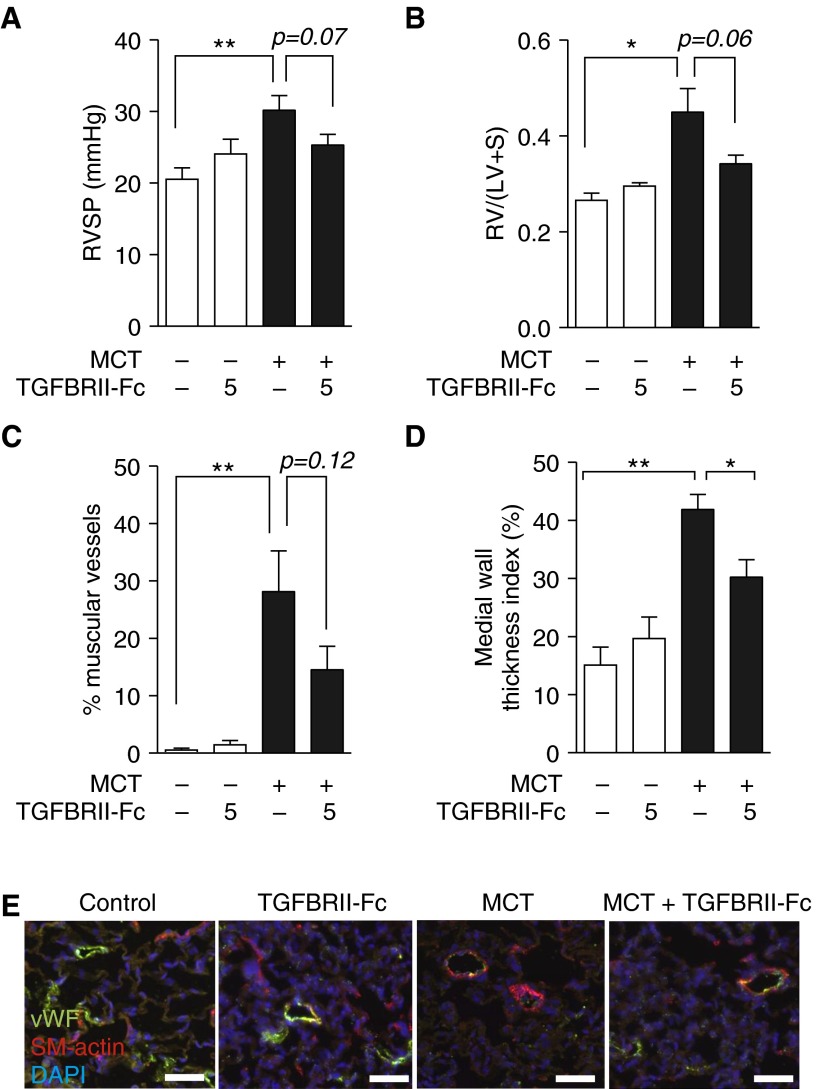
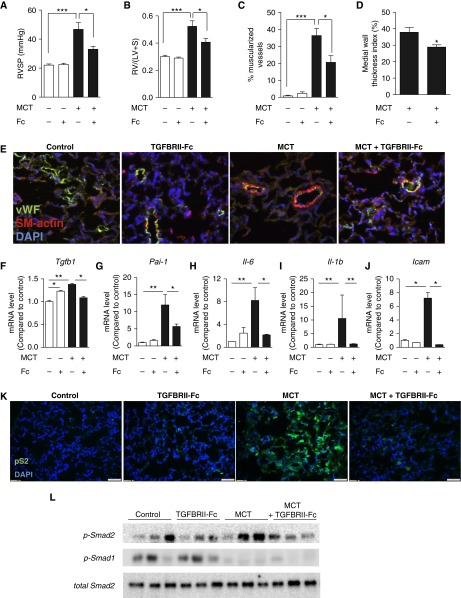
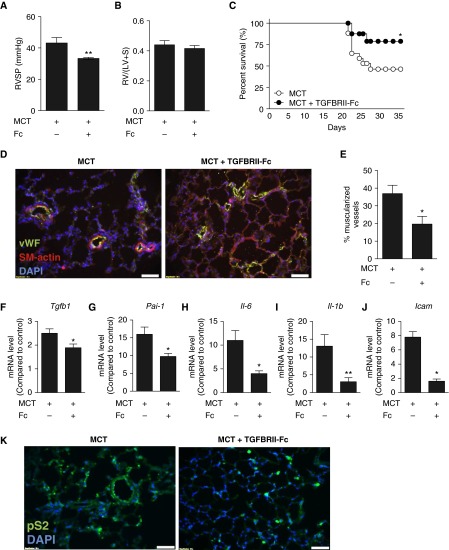
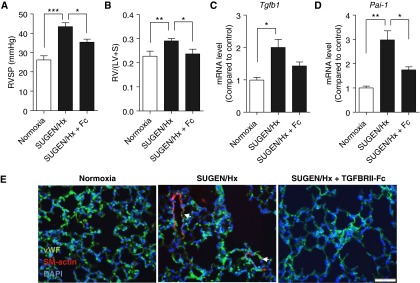
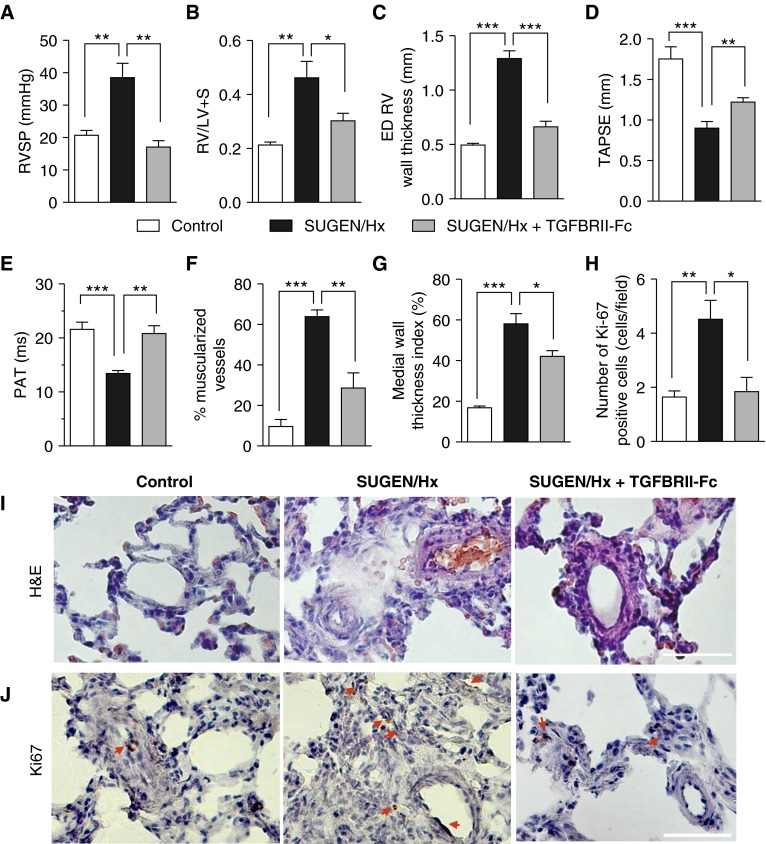
Measurements and main results: TGFBRII-Fc is an inhibitor of TGF-β1 and TGF-β3, but not TGF-β2, signaling. In vivo treatment with TGFBRII-Fc attenuated Smad2 phosphorylation, normalized expression of plasminogen activator inhibitor-1, and mitigated PH and pulmonary vascular remodeling in monocrotaline-treated rats, SU5416/hypoxia-treated rats, and SU5416/hypoxia-treated mice. Administration of TGFBRII-Fc to monocrotaline-treated or SU5416/hypoxia-treated rats with established PH improved right ventricular systolic pressures, right ventricular function, and survival. No cardiac structural or valvular abnormalities were observed after treatment with TGFBRII-Fc.
Conclusions: Our findings are consistent with a pathogenetic role of TGF-β1/3, demonstrating the efficacy and tolerability of selective TGF-β ligand blockade for improving hemodynamics, remodeling, and survival in multiple experimental PH models.
Keywords: pulmonary artery; pulmonary hypertension; transforming growth factor-β; vascular remodeling; vascular smooth muscle cells.
Figures
Comment in
-
Catching a Disease: A Molecular Trap as a Therapy for Pulmonary Arterial Hypertension.Am J Respir Crit Care Med. 2016 Nov 1;194(9):1047-1049. doi: 10.1164/rccm.201605-0920ED. Am J Respir Crit Care Med. 2016. PMID: 27797615 Free PMC article. No abstract available.
References
-
- Strelau J, Schober A, Sullivan A, Schilling L, Unsicker K. Growth/differentiation factor-15 (GDF-15), a novel member of the TGF-beta superfamily, promotes survival of lesioned mesencephalic dopaminergic neurons in vitro and in vivo and is induced in neurons following cortical lesioning. J Neural Transm Suppl. 2003;(65):197–203. - PubMed
-
- Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, III, Loyd JE, Nichols WC, Trembath RC International PPH Consortium. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26:81–84. - PubMed
-
- Waite KA, Eng C. From developmental disorder to heritable cancer: it’s all in the BMP/TGF-beta family. Nat Rev Genet. 2003;4:763–773. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
