

Differential utilization of binding loop flexibility in T cell receptor ligand selection and cross-reactivity
- PMID: 27118724
- PMCID: PMC4846865
- DOI: 10.1038/srep25070
Differential utilization of binding loop flexibility in T cell receptor ligand selection and cross-reactivity
Abstract
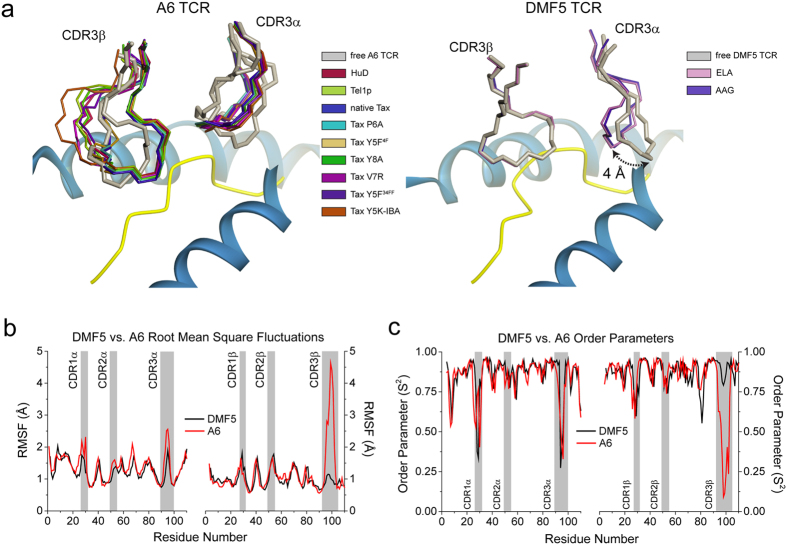
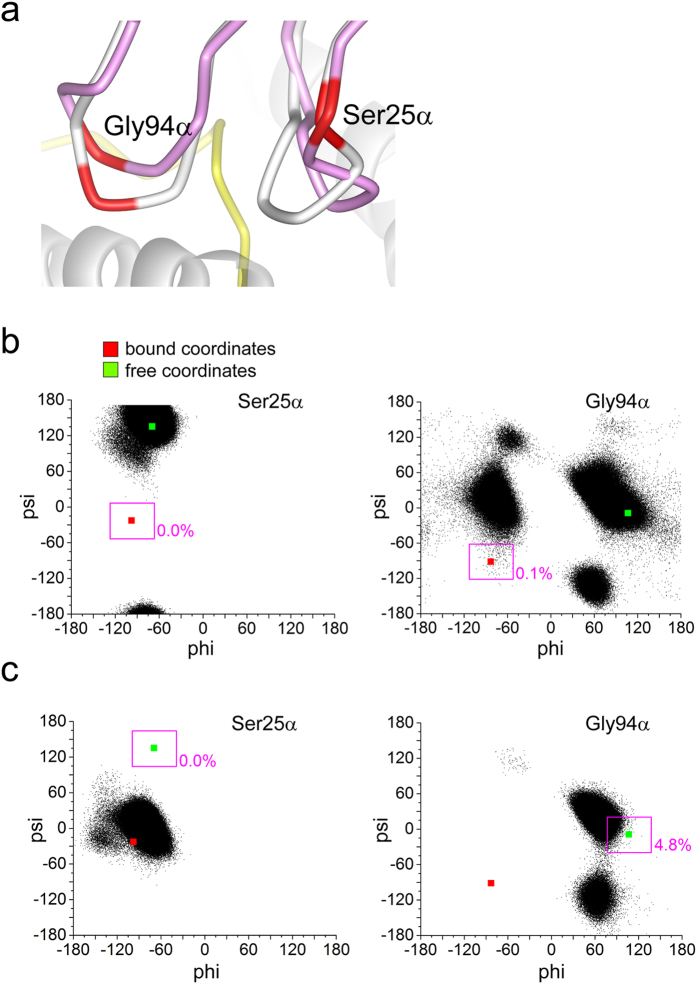
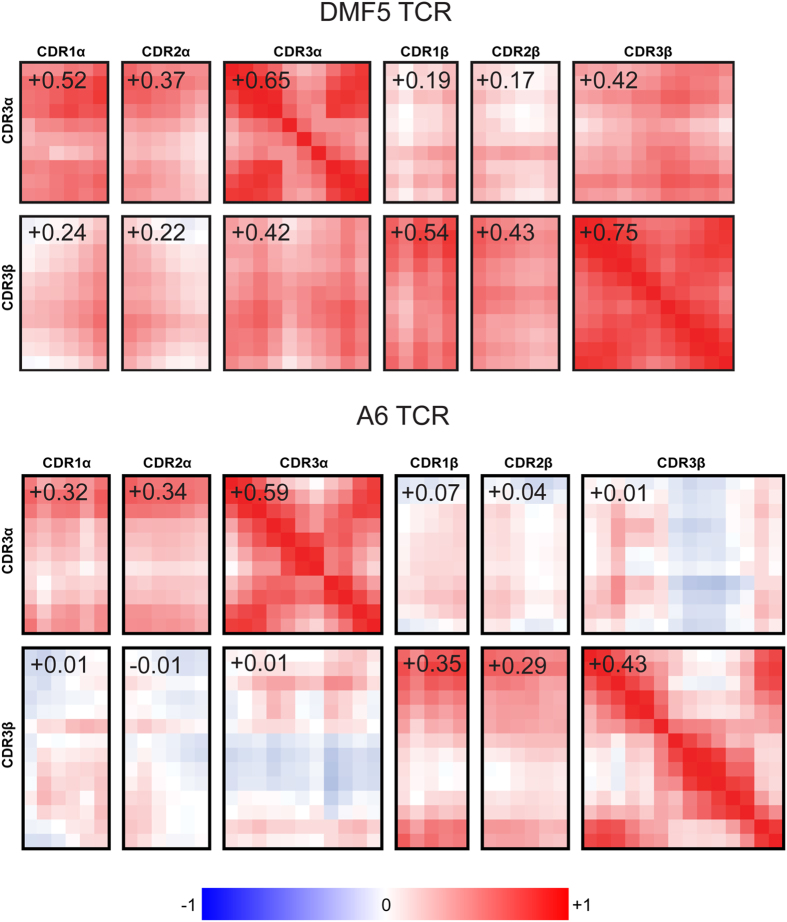
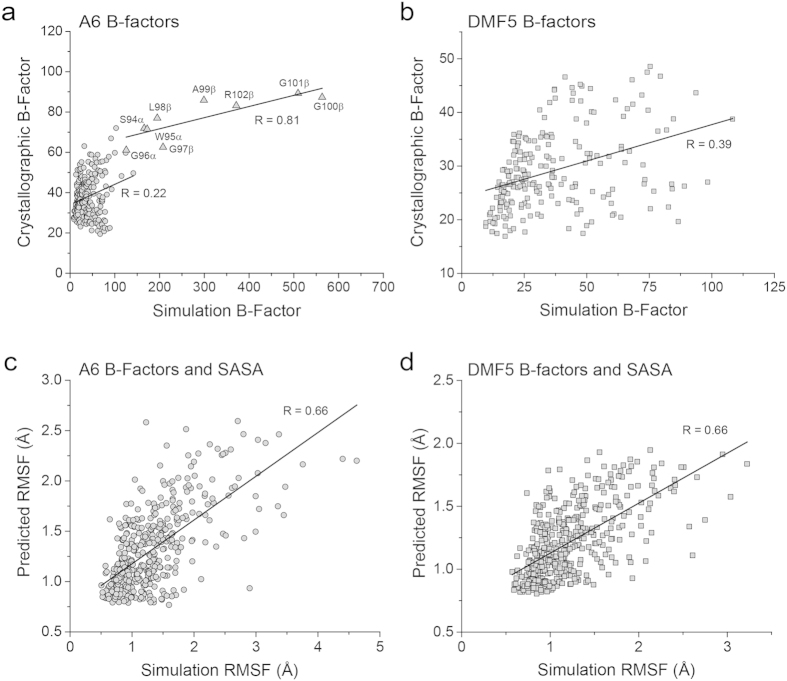
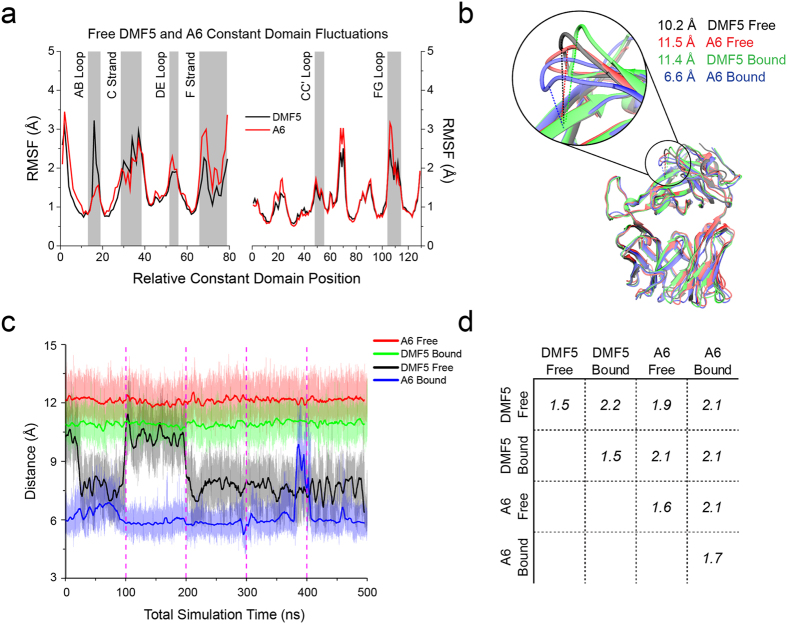
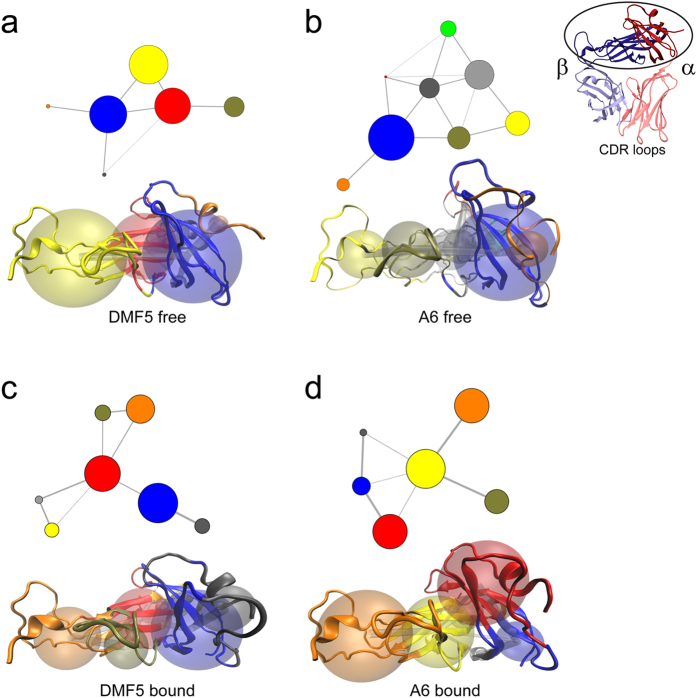
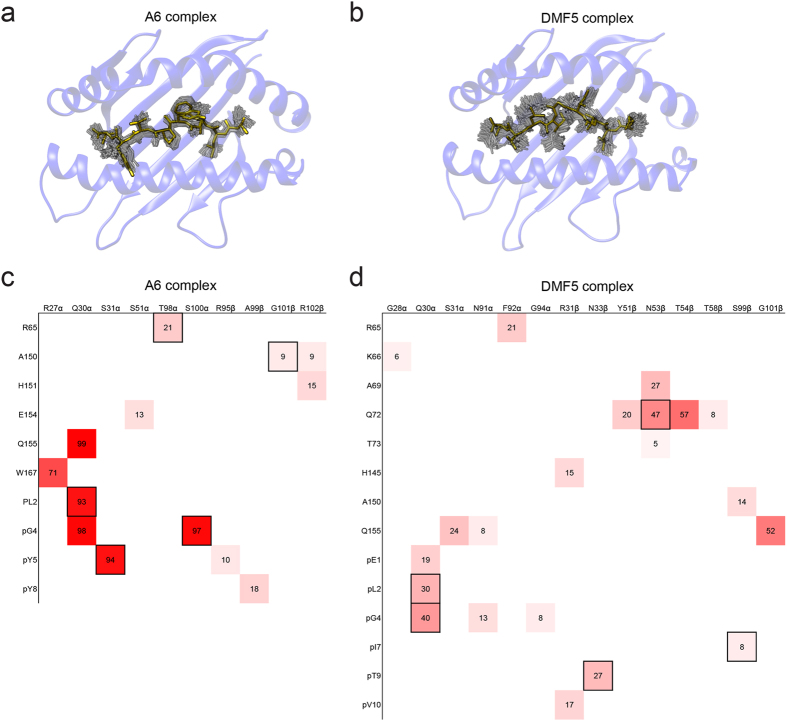
Complementarity determining region (CDR) loop flexibility has been suggested to play an important role in the selection and binding of ligands by T cell receptors (TCRs) of the cellular immune system. However, questions remain regarding the role of loop motion in TCR binding, and crystallographic structures have raised questions about the extent to which generalizations can be made. Here we studied the flexibility of two structurally well characterized αβ TCRs, A6 and DMF5. We found that the two receptors utilize loop motion very differently in ligand binding and cross-reactivity. While the loops of A6 move rapidly in an uncorrelated fashion, those of DMF5 are substantially less mobile. Accordingly, the mechanisms of binding and cross-reactivity are very different between the two TCRs: whereas A6 relies on conformational selection to select and bind different ligands, DMF5 uses a more rigid, permissive architecture with greater reliance on slower motions or induced-fit. In addition to binding site flexibility, we also explored whether ligand-binding resulted in common dynamical changes in A6 and DMF5 that could contribute to TCR triggering. Although binding-linked motional changes propagated throughout both receptors, no common features were observed, suggesting that changes in nanosecond-level TCR structural dynamics do not contribute to T cell signaling.
Figures
References
-
- Mason D. A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunology Today 19, 395–404 (1998). - PubMed
-
- Rossjohn J. et al. T Cell Antigen Receptor Recognition of Antigen-Presenting Molecules. Annual Review of Immunology 33, 169–200 (2015). - PubMed
-
- Gagnon S. J. et al. T Cell Receptor Recognition via Cooperative Conformational Plasticity. Journal of Molecular Biology 363, 228–243 (2006). - PubMed
-
- Garcia K. C. et al. Structural basis of plasticity in T cell receptor recognition of a self peptide-MHC antigen. Science 279, 1166–1172 (1998). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
