

Traumatic brain injury: pathophysiology for neurocritical care
- PMID: 27123305
- PMCID: PMC4847183
- DOI: 10.1186/s40560-016-0138-3
Traumatic brain injury: pathophysiology for neurocritical care
Abstract
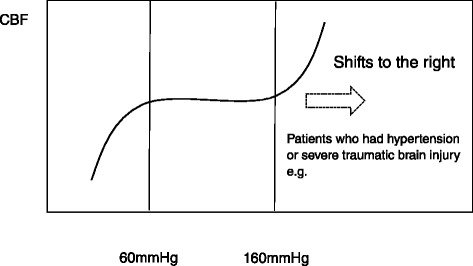
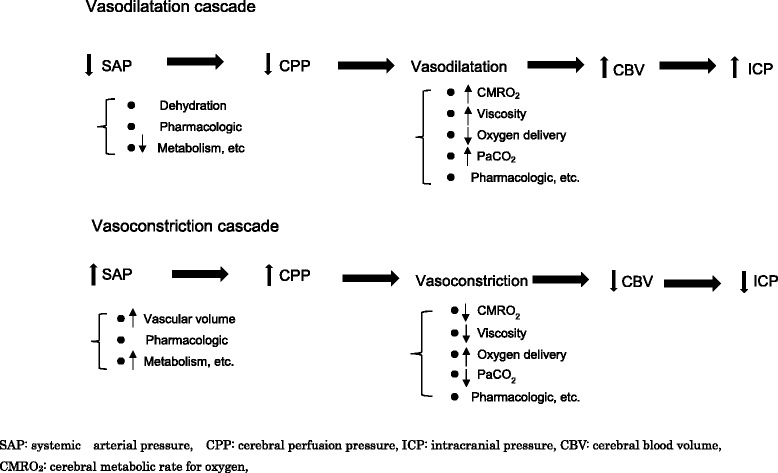
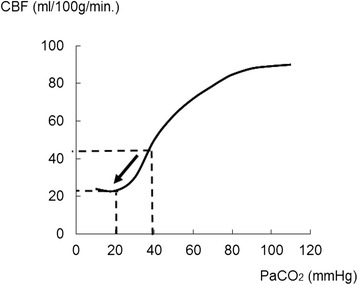
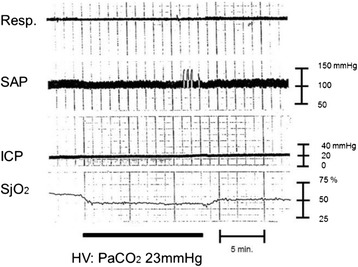
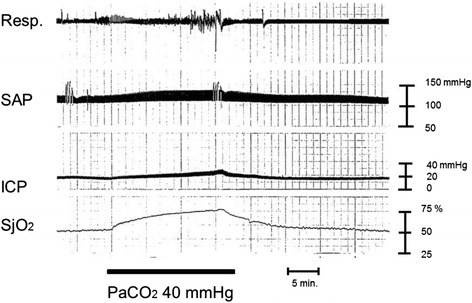
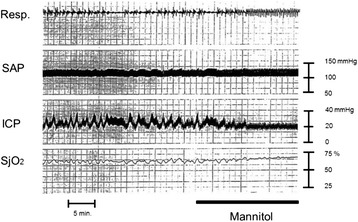
Severe cases of traumatic brain injury (TBI) require neurocritical care, the goal being to stabilize hemodynamics and systemic oxygenation to prevent secondary brain injury. It is reported that approximately 45 % of dysoxygenation episodes during critical care have both extracranial and intracranial causes, such as intracranial hypertension and brain edema. For this reason, neurocritical care is incomplete if it only focuses on prevention of increased intracranial pressure (ICP) or decreased cerebral perfusion pressure (CPP). Arterial hypotension is a major risk factor for secondary brain injury, but hypertension with a loss of autoregulation response or excess hyperventilation to reduce ICP can also result in a critical condition in the brain and is associated with a poor outcome after TBI. Moreover, brain injury itself stimulates systemic inflammation, leading to increased permeability of the blood-brain barrier, exacerbated by secondary brain injury and resulting in increased ICP. Indeed, systemic inflammatory response syndrome after TBI reflects the extent of tissue damage at onset and predicts further tissue disruption, producing a worsening clinical condition and ultimately a poor outcome. Elevation of blood catecholamine levels after severe brain damage has been reported to contribute to the regulation of the cytokine network, but this phenomenon is a systemic protective response against systemic insults. Catecholamines are directly involved in the regulation of cytokines, and elevated levels appear to influence the immune system during stress. Medical complications are the leading cause of late morbidity and mortality in many types of brain damage. Neurocritical care after severe TBI has therefore been refined to focus not only on secondary brain injury but also on systemic organ damage after excitation of sympathetic nerves following a stress reaction.
Keywords: Catecholamine; Hyperglycemia; Neurocritical care; Pathophysiology; Traumatic brain injury.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
