

Brain injury following cardiac arrest: pathophysiology for neurocritical care
- PMID: 27123307
- PMCID: PMC4847238
- DOI: 10.1186/s40560-016-0140-9
Brain injury following cardiac arrest: pathophysiology for neurocritical care
Retraction in
-
Retraction Note: Brain injury following cardiac arrest: pathophysiology for neurocritical care.J Intensive Care. 2016 Sep 20;4:61. doi: 10.1186/s40560-016-0185-9. eCollection 2016. J Intensive Care. 2016. PMID: 27672440 Free PMC article.
Abstract
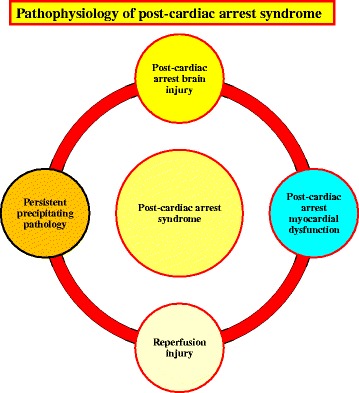
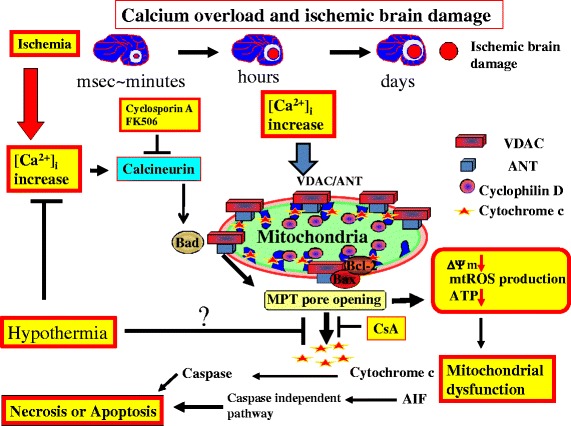
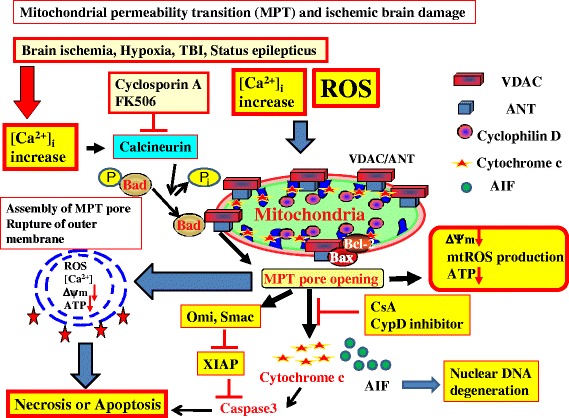
Cardiac arrest induces the cessation of cerebral blood flow, which can result in brain damage. The primary intervention to salvage the brain under such a pathological condition is to restore the cerebral blood flow to the ischemic region. Ischemia is defined as a reduction in blood flow to a level that is sufficient to alter normal cellular function. Brain tissue is highly sensitive to ischemia, such that even brief ischemic periods in neurons can initiate a complex sequence of events that may ultimately culminate in cell death. However, paradoxically, restoration of blood flow can cause additional damage and exacerbate the neurocognitive deficits in patients who suffered a brain ischemic event, which is a phenomenon referred to as "reperfusion injury." Transient brain ischemia following cardiac arrest results from the complex interplay of multiple pathways including excitotoxicity, acidotoxicity, ionic imbalance, peri-infarct depolarization, oxidative and nitrative stress, inflammation, and apoptosis. The pathophysiology of post-cardiac arrest brain injury involves a complex cascade of molecular events, most of which remain unknown. Many lines of evidence have shown that mitochondria suffer severe damage in response to ischemic injury. Mitochondrial dysfunction based on the mitochondrial permeability transition after reperfusion, particularly involving the calcineurin/immunophilin signal transduction pathway, appears to play a pivotal role in the induction of neuronal cell death. The aim of this article is to discuss the underlying pathophysiology of brain damage, which is a devastating pathological condition, and highlight the central signal transduction pathway involved in brain damage, which reveals potential targets for therapeutic intervention.
Keywords: Calcineurin/immunophilin; Cardiac arrest; Excitotoxicity; Mitochondrial dysfunction; Mitochondrial permeability transition (MPT); Pathophysiology of ischemic brain damage; Post-cardiac arrest syndrome (PCAS); Reperfusion injury.
Figures

References
-
- Peberdy MA, Callaway CW, Neumar RW, Geocadin RG, Zimmerman JL, Donnino M, et al. Part 9: post-cardiac arrest care: 2010 American Heart Association guidelines for cardiopulmonary resuscitation and emergency cardiovascular care. Circulation. 2010;122:S768–86. - PubMed
-
- Uchino H, Kuroda Y, Morota S, Hirabayashi G, Ishii N, Shibasaki F, Ikeda Y, Hansson MJ, Elmér E.Probing the molecular mechanisms of neuronal degeneration: importance of mitochondrial dysfunction andcalcineurin activation. J Anesth. 2008;22(3):253-62. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
