

Porphyromonas gingivalis Differentially Modulates Cell Death Profile in Ox-LDL and TNF-α Pre-Treated Endothelial Cells
- PMID: 27124409
- PMCID: PMC4849801
- DOI: 10.1371/journal.pone.0154590
Porphyromonas gingivalis Differentially Modulates Cell Death Profile in Ox-LDL and TNF-α Pre-Treated Endothelial Cells
Abstract
Objective: Clinical studies demonstrated a potential link between atherosclerosis and periodontitis. Porphyromonas gingivalis (Pg), one of the main periodontal pathogen, has been associated to atheromatous plaque worsening. However, synergism between infection and other endothelial stressors such as oxidized-LDL or TNF-α especially on endothelial cell (EC) death has not been investigated. This study aims to assess the role of Pg on EC death in an inflammatory context and to determine potential molecular pathways involved.
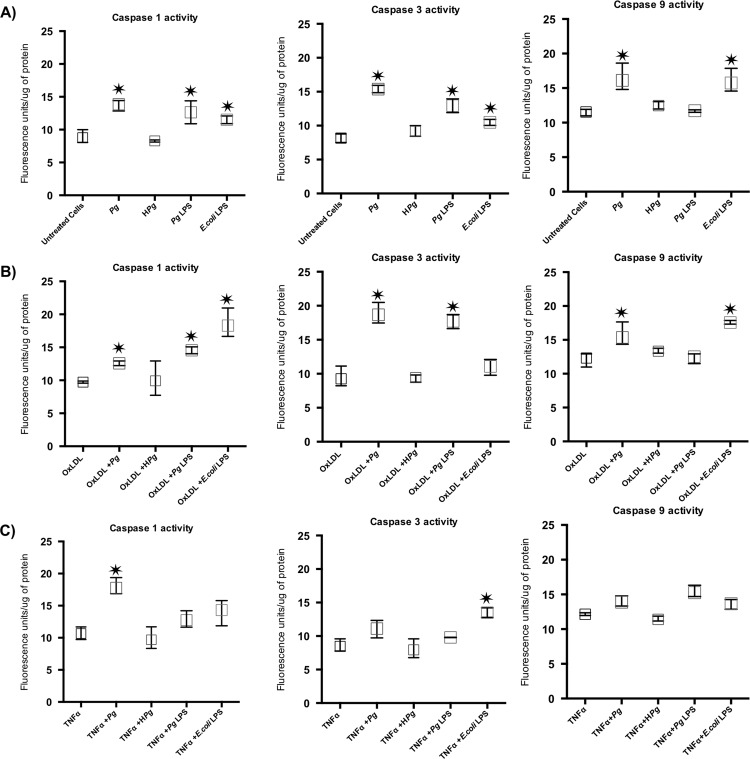
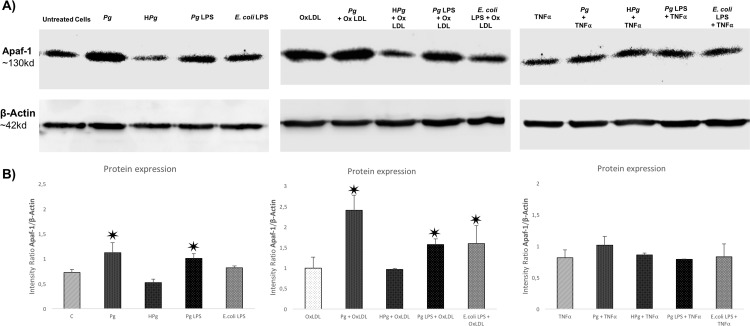
Methods: Human umbilical vein ECs (HUVECs) were infected with Pg (MOI 100) or stimulated by its lipopolysaccharide (Pg-LPS) (1μg/ml) for 24 to 48 hours. Cell viability was measured with AlamarBlue test, type of cell death induced was assessed using Annexin V/propidium iodide staining. mRNA expression regarding caspase-1, -3, -9, Bcl-2, Bax-1 and Apaf-1 has been evaluated with RT-qPCR. Caspases enzymatic activity and concentration of APAF-1 protein were evaluated to confirm mRNA results.
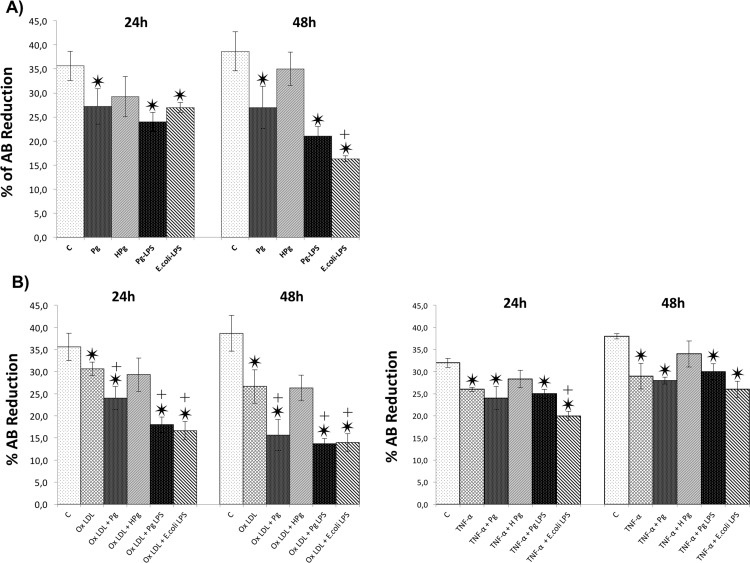
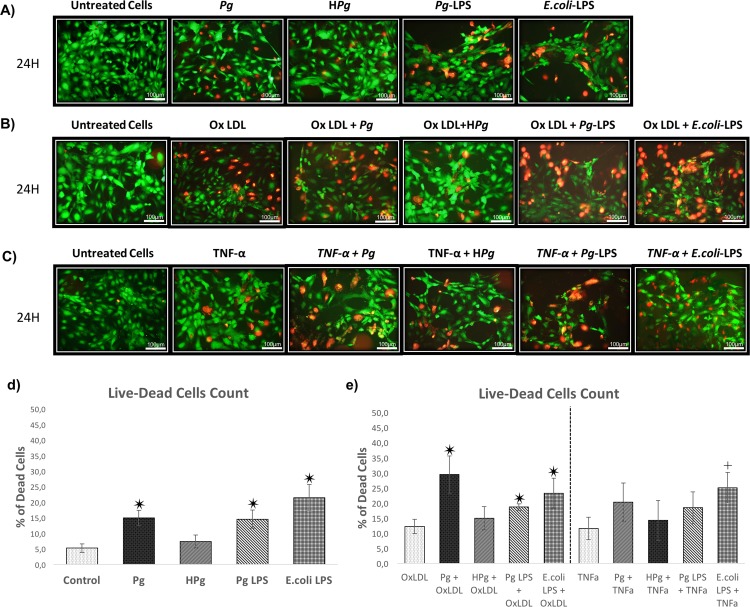
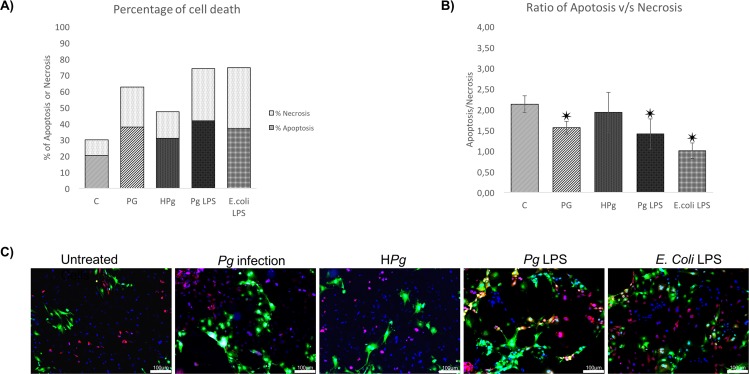
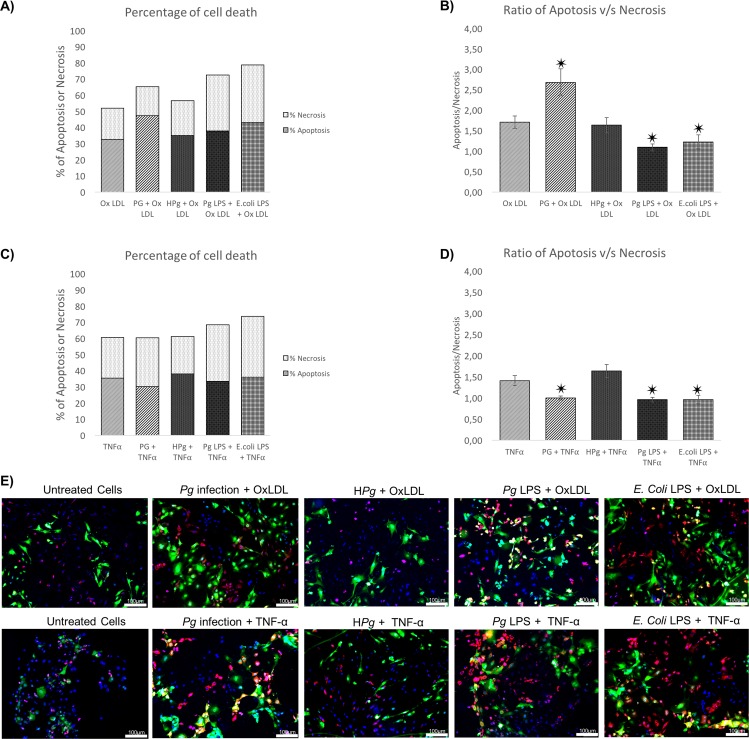
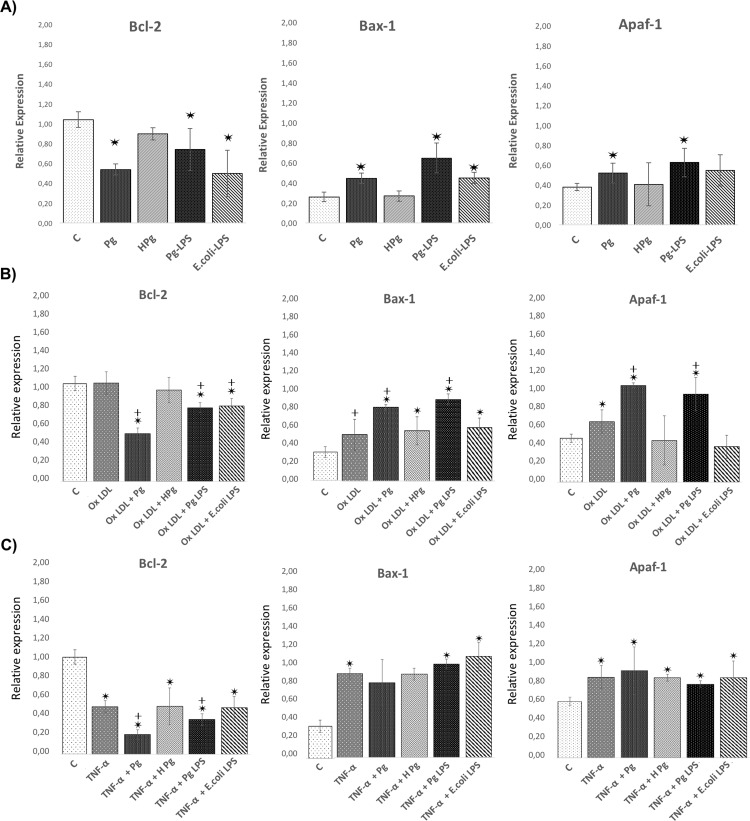
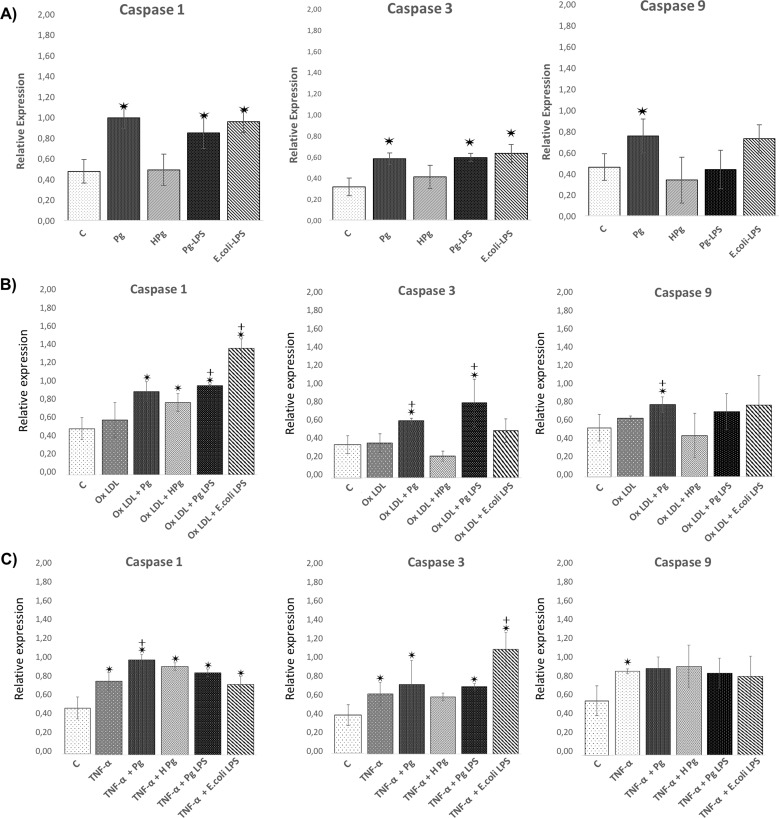
Results: Pg infection and Pg-LPS stimulation induced EC death. A cumulative effect has been observed in Ox-LDL pre-treated ECs infected or stimulated. This effect was not observed in TNF-α pre-treated cells. Pg infection promotes EC necrosis, however, in infected Ox-LDL pre-treated ECs, apoptosis was promoted. This effect was not observed in TNF-α pre-treated cells highlighting specificity of molecular pathways activated. Regarding mRNA expression, Pg increased expression of pro-apoptotic genes including caspases-1,-3,-9, Bax-1 and decreased expression of anti-apoptotic Bcl-2. In Ox-LDL pre-treated ECs, Pg increased significantly the expression of Apaf-1. These results were confirmed at the protein level.
Conclusion: This study contributes to demonstrate that Pg and its Pg-LPS could exacerbate Ox-LDL and TNF-α induced endothelial injury through increase of EC death. Interestingly, molecular pathways are differentially modulated by the infection in function of the pre-stimulation.
Conflict of interest statement
Figures
References
-
- Lockhart PB, Bolger AF, Papapanou PN, Osinbowale O, Trevisan M, Levison ME, et al. Periodontal Disease and Atherosclerotic Vascular Disease: Does the Evidence Support an Independent Association?: A Scientific Statement From the American Heart Association. Circulation. 2012;125: 2520–2544. 10.1161/CIR.0b013e31825719f3 - DOI - PubMed
-
- Huck O, Saadi-Thiers K, Tenenbaum H, Davideau J-L, Romagna C, Laurent Y, et al. Evaluating periodontal risk for patients at risk of or suffering from atherosclerosis: recent biological hypotheses and therapeutic consequences. Archives of Cardiovascular Diseases. 2011;104: 352–358. 10.1016/j.acvd.2011.02.002 - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
