

RNA splicing is a primary link between genetic variation and disease
- PMID: 27126046
- PMCID: PMC5182069
- DOI: 10.1126/science.aad9417
RNA splicing is a primary link between genetic variation and disease
Abstract
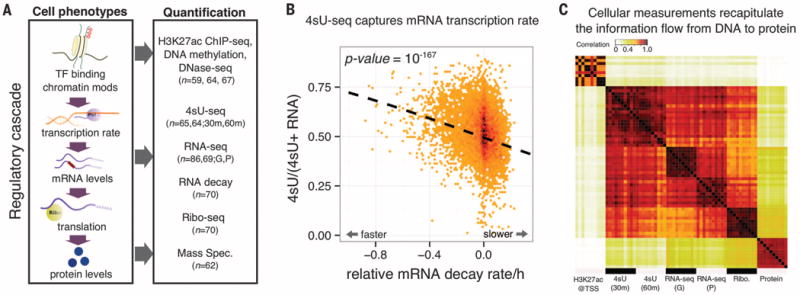
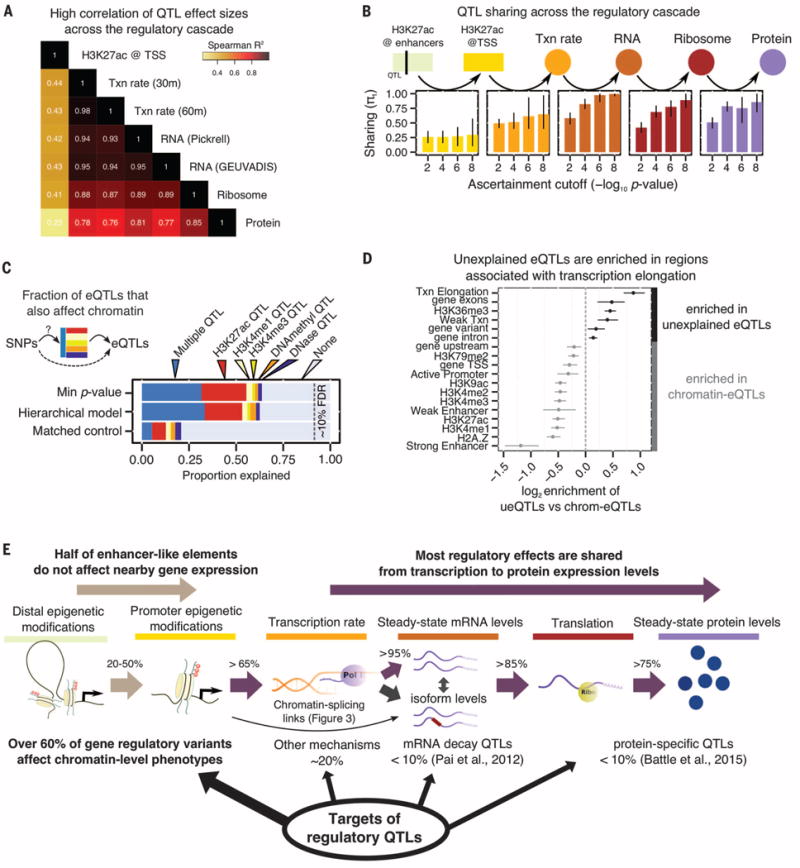
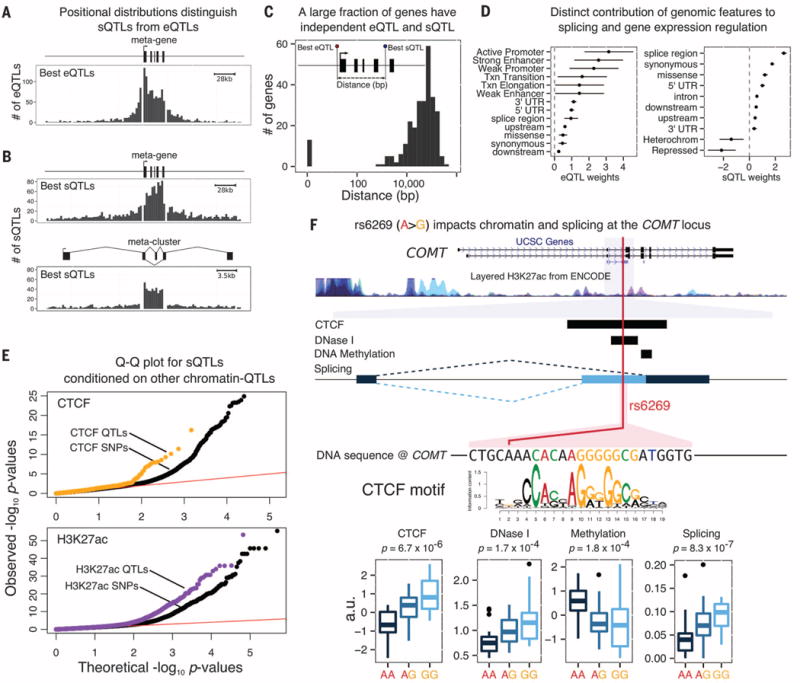
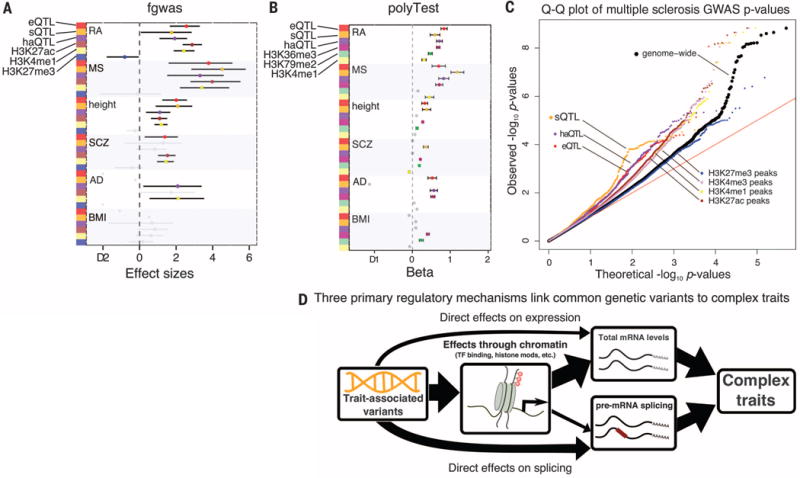
Noncoding variants play a central role in the genetics of complex traits, but we still lack a full understanding of the molecular pathways through which they act. We quantified the contribution of cis-acting genetic effects at all major stages of gene regulation from chromatin to proteins, in Yoruba lymphoblastoid cell lines (LCLs). About ~65% of expression quantitative trait loci (eQTLs) have primary effects on chromatin, whereas the remaining eQTLs are enriched in transcribed regions. Using a novel method, we also detected 2893 splicing QTLs, most of which have little or no effect on gene-level expression. These splicing QTLs are major contributors to complex traits, roughly on a par with variants that affect gene expression levels. Our study provides a comprehensive view of the mechanisms linking genetic variation to variation in human gene regulation.
Copyright © 2016, American Association for the Advancement of Science.
Figures
Comment in
-
Human genetics: Splicing: linking genetic variation and disease.Nat Rev Genet. 2016 Jun;17(6):317. doi: 10.1038/nrg.2016.64. Epub 2016 May 9. Nat Rev Genet. 2016. PMID: 27156973 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
