

Targeting TDP-43 phosphorylation by Casein Kinase-1δ inhibitors: a novel strategy for the treatment of frontotemporal dementia
- PMID: 27138926
- PMCID: PMC4852436
- DOI: 10.1186/s13024-016-0102-7
Targeting TDP-43 phosphorylation by Casein Kinase-1δ inhibitors: a novel strategy for the treatment of frontotemporal dementia
Abstract
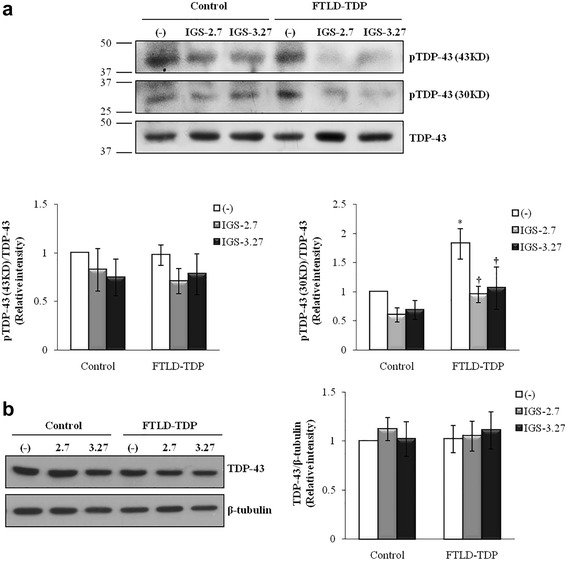
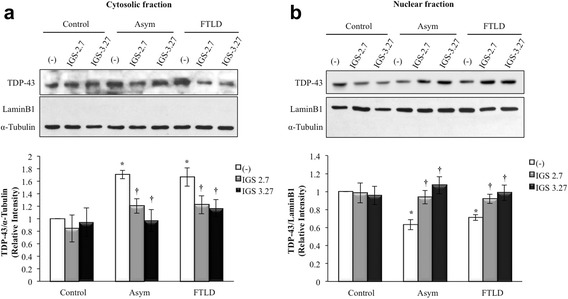
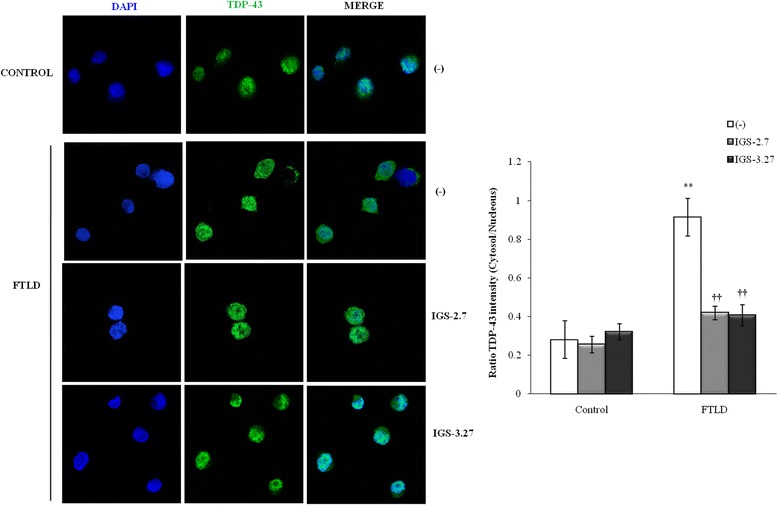
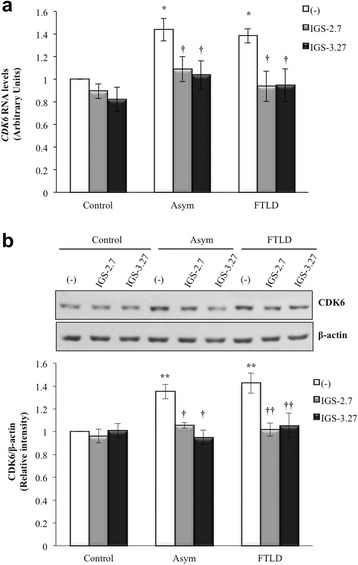
Background: Mutations in the progranulin gene (GRN) are the most common cause of frontotemporal lobar degeneration with TDP-43 inclusions (FTLD-TDP). TDP-43 pathology is characterized by the hyperphosphorylation of the protein at Serine 409/410 residues. Casein kinase-1δ (CK-1δ) was reported to phosphorylate TDP-43 directly. Previous works from our laboratory described the presence of CDK6/pRb-dependent cell cycle alterations, and cytosolic accumulation of TDP-43 protein in lymphoblast from FTLD-TDP patients carriers of a loss-of function mutation in GRN gene (c.709-1G > A). In this work, we have investigated the effects of two brain penetrant CK-1δ inhibitors (IGS-2.7 and IGS-3.27) designed and synthetized in our laboratory on cell proliferation, TDP-43 phosphorylation and subcellular localization, as well as their effects on the known nuclear TDP-43 function repressing the expression of CDK6.
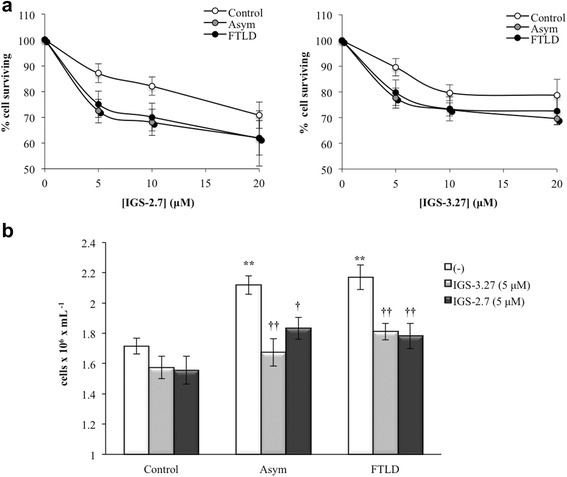
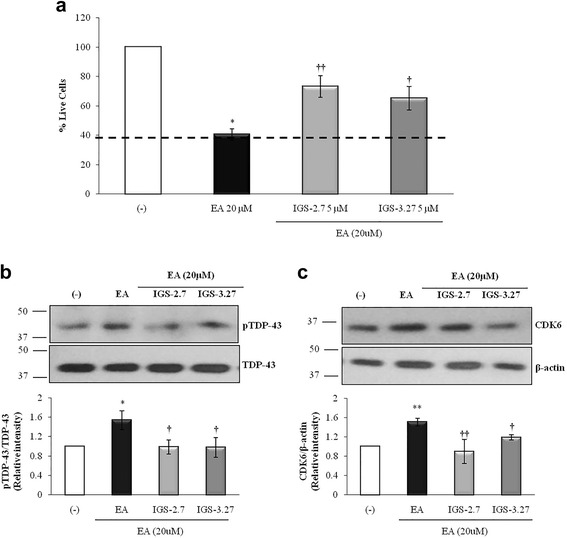
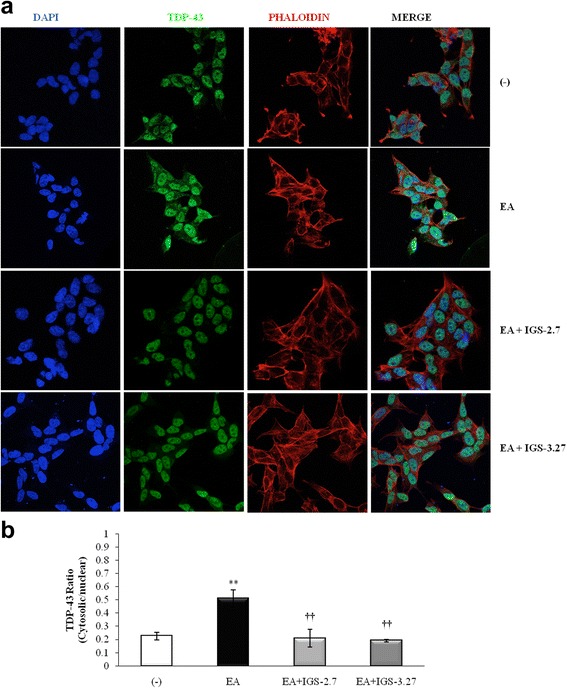
Results: We report here that both CK-1δ inhibitors (IGS-2.7 and IGS-3.27) normalized the proliferative activity of PGRN-deficient lymphoblasts by preventing the phosphorylation of TDP-43 fragments, its nucleo-cytosol translocation and the overactivation of the CDK6/pRb cascade. Moreover, ours results show neuroprotective effects of CK-1δ inhibitors in a neuronal cell model of induced TDP-43 phosphorylation.
Conclusions: Our results suggest that modulating CK-1δ activity could be considered a novel therapeutic approach for the treatment of FTLD-TDP and other TDP-43 proteinopathies.
Keywords: CDK6; CK-1δ; Cell proliferation; FTLD-TDP; Lymphocytes; TDP-43.
Figures

References
-
- Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–4. doi: 10.1038/nature05017. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
