

Using iterative fragment assembly and progressive sequence truncation to facilitate phasing and crystal structure determination of distantly related proteins
- PMID: 27139625
- PMCID: PMC4931812
- DOI: 10.1107/S2059798316003016
Using iterative fragment assembly and progressive sequence truncation to facilitate phasing and crystal structure determination of distantly related proteins
Abstract
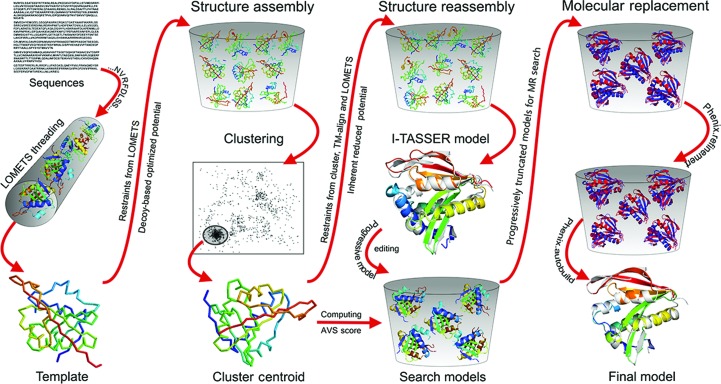
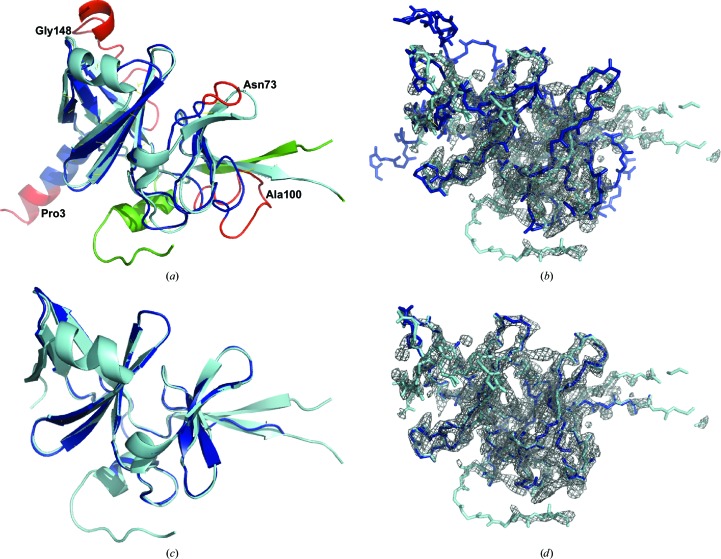
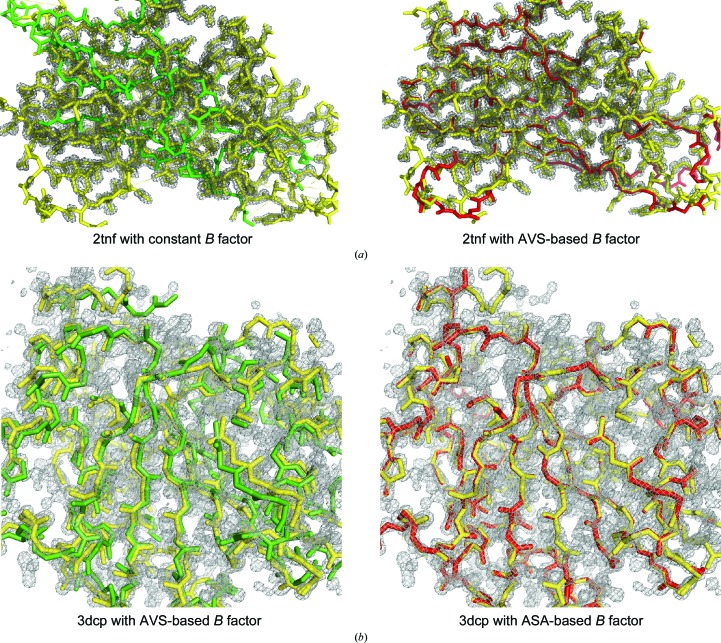
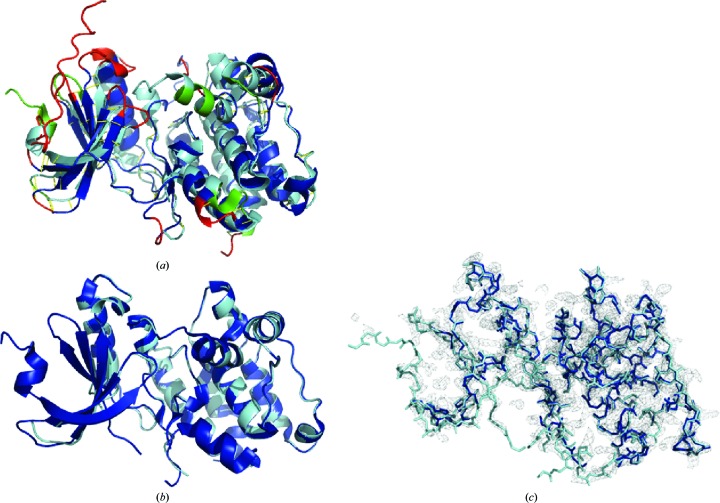
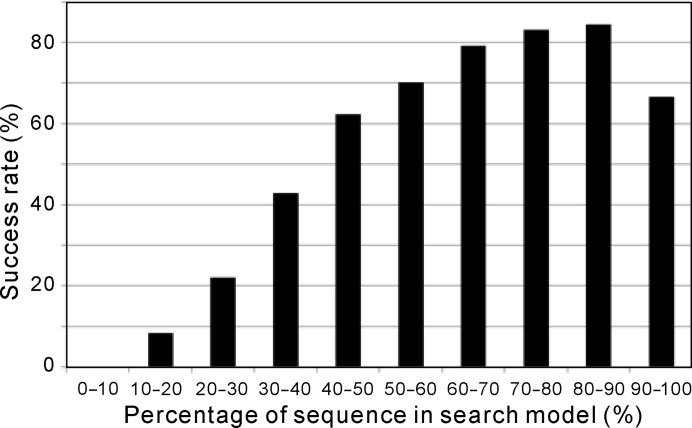
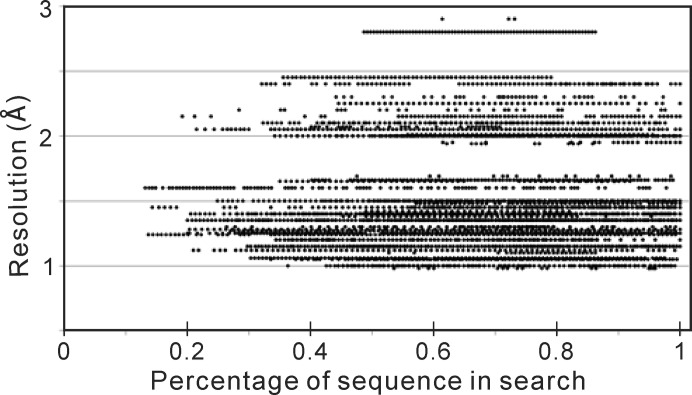
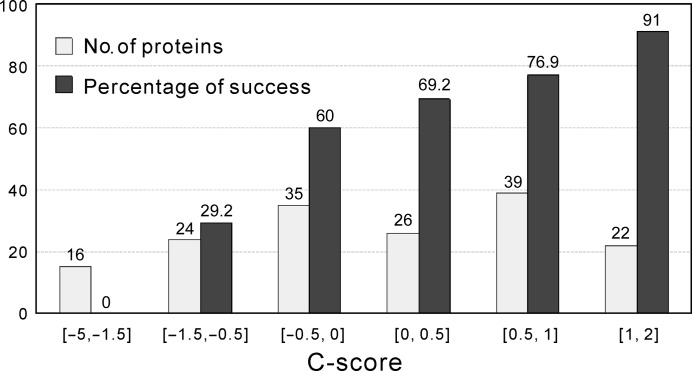
Molecular replacement (MR) often requires templates with high homology to solve the phase problem in X-ray crystallography. I-TASSER-MR has been developed to test whether the success rate for structure determination of distant-homology proteins could be improved by a combination of iterative fragmental structure-assembly simulations with progressive sequence truncation designed to trim regions with high variation. The pipeline was tested on two independent protein sets consisting of 61 proteins from CASP8 and 100 high-resolution proteins from the PDB. After excluding homologous templates, I-TASSER generated full-length models with an average TM-score of 0.773, which is 12% higher than the best threading templates. Using these as search models, I-TASSER-MR found correct MR solutions for 95 of 161 targets as judged by having a TFZ of >8 or with the final structure closer to the native than the initial search models. The success rate was 16% higher than when using the best threading templates. I-TASSER-MR was also applied to 14 protein targets from structure genomics centers. Seven of these were successfully solved by I-TASSER-MR. These results confirm that advanced structure assembly and progressive structural editing can significantly improve the success rate of MR for targets with distant homology to proteins of known structure.
Keywords: I-TASSER; X-ray crystallography; molecular replacement; protein structure prediction; threading.
Figures
Similar articles
-
I-TASSER-MR: automated molecular replacement for distant-homology proteins using iterative fragment assembly and progressive sequence truncation.Nucleic Acids Res. 2017 Jul 3;45(W1):W429-W434. doi: 10.1093/nar/gkx349. Nucleic Acids Res. 2017. PMID: 28472524 Free PMC article.
-
Template-based protein structure prediction in CASP11 and retrospect of I-TASSER in the last decade.Proteins. 2016 Sep;84 Suppl 1(Suppl 1):233-46. doi: 10.1002/prot.24918. Epub 2015 Sep 18. Proteins. 2016. PMID: 26343917 Free PMC article.
-
NMR data-driven structure determination using NMR-I-TASSER in the CASD-NMR experiment.J Biomol NMR. 2015 Aug;62(4):511-25. doi: 10.1007/s10858-015-9914-y. Epub 2015 Mar 4. J Biomol NMR. 2015. PMID: 25737244 Free PMC article.
-
I-TASSER-MTD: a deep-learning-based platform for multi-domain protein structure and function prediction.Nat Protoc. 2022 Oct;17(10):2326-2353. doi: 10.1038/s41596-022-00728-0. Epub 2022 Aug 5. Nat Protoc. 2022. PMID: 35931779 Review.
-
Applications of ACORN to data at 1.45 A resolution.J Synchrotron Radiat. 2004 Jan 1;11(Pt 1):64-7. doi: 10.1107/s0909049503023537. Epub 2003 Nov 28. J Synchrotron Radiat. 2004. PMID: 14646136 Review.
Cited by
-
MR-REX: molecular replacement by cooperative conformational search and occupancy optimization on low-accuracy protein models.Acta Crystallogr D Struct Biol. 2018 Jul 1;74(Pt 7):606-620. doi: 10.1107/S2059798318005612. Epub 2018 Jun 8. Acta Crystallogr D Struct Biol. 2018. PMID: 29968671 Free PMC article.
-
Approaches to ab initio molecular replacement of α-helical transmembrane proteins.Acta Crystallogr D Struct Biol. 2017 Dec 1;73(Pt 12):985-996. doi: 10.1107/S2059798317016436. Epub 2017 Nov 22. Acta Crystallogr D Struct Biol. 2017. PMID: 29199978 Free PMC article.
-
Fragon: rapid high-resolution structure determination from ideal protein fragments.Acta Crystallogr D Struct Biol. 2018 Mar 1;74(Pt 3):205-214. doi: 10.1107/S2059798318002292. Epub 2018 Mar 2. Acta Crystallogr D Struct Biol. 2018. PMID: 29533228 Free PMC article.
-
I-TASSER-MR: automated molecular replacement for distant-homology proteins using iterative fragment assembly and progressive sequence truncation.Nucleic Acids Res. 2017 Jul 3;45(W1):W429-W434. doi: 10.1093/nar/gkx349. Nucleic Acids Res. 2017. PMID: 28472524 Free PMC article.
-
Molecular-replacement phasing using predicted protein structures from AWSEM-Suite.IUCrJ. 2020 Oct 27;7(Pt 6):1168-1178. doi: 10.1107/S2052252520013494. eCollection 2020 Nov 1. IUCrJ. 2020. PMID: 33209327 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
