

Developmental signalling pathways in renal fibrosis: the roles of Notch, Wnt and Hedgehog
- PMID: 27140856
- PMCID: PMC5529143
- DOI: 10.1038/nrneph.2016.54
Developmental signalling pathways in renal fibrosis: the roles of Notch, Wnt and Hedgehog
Abstract
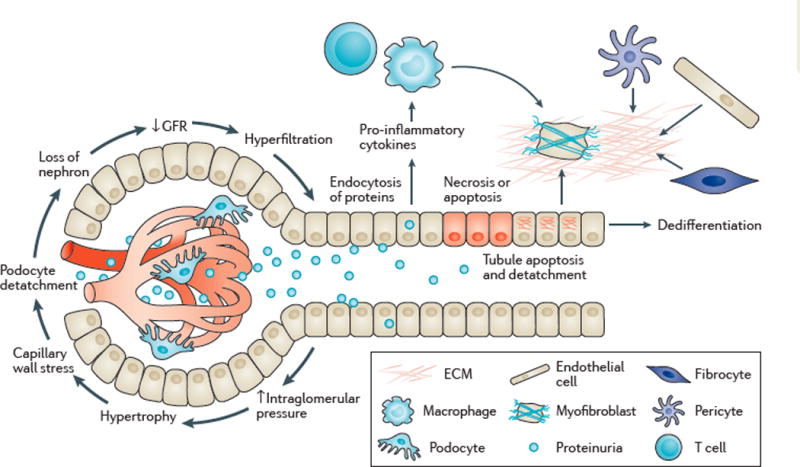
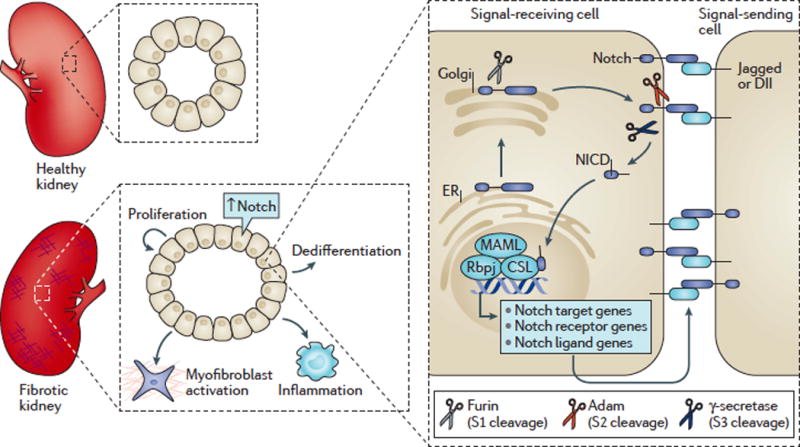
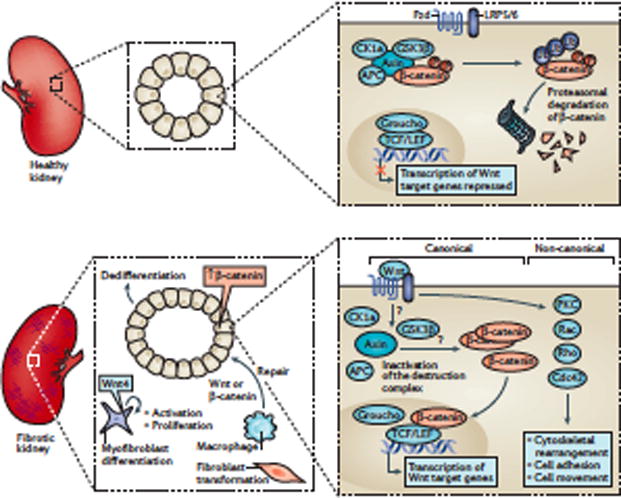
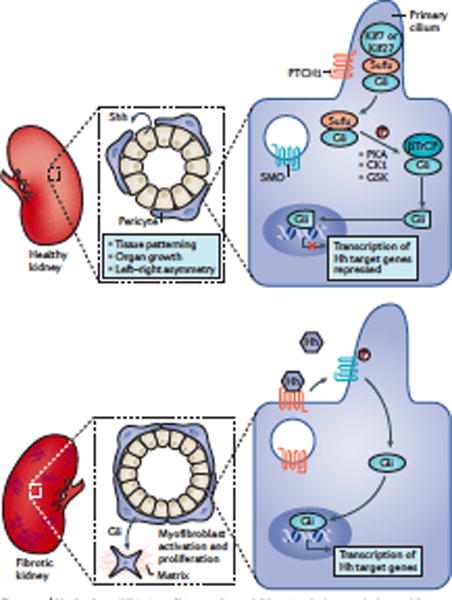
Kidney fibrosis is a common histological manifestation of functional decline in the kidney. Fibrosis is a reactive process that develops in response to excessive epithelial injury and inflammation, leading to myofibroblast activation and an accumulation of extracellular matrix. Here, we describe how three key developmental signalling pathways - Notch, Wnt and Hedgehog (Hh) - are reactivated in response to kidney injury and contribute to the fibrotic response. Although transient activation of these pathways is needed for repair of injured tissue, their sustained activation is thought to promote fibrosis. Excessive Wnt and Notch expression prohibit epithelial differentiation, whereas increased Wnt and Hh expression induce fibroblast proliferation and myofibroblastic transdifferentiation. Notch, Wnt and Hh are fundamentally different signalling pathways, but their choreographed activation seems to be just as important for fibrosis as it is for embryonic kidney development. Decreasing the activity of Notch, Wnt or Hh signalling could potentially provide a new therapeutic strategy to ameliorate the development of fibrosis in chronic kidney disease.
Conflict of interest statement
The Susztaklab received research support from Boehringer Ingelheim, Biogen and Lilly.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
