

An evaluation of methods correcting for cell-type heterogeneity in DNA methylation studies
- PMID: 27142380
- PMCID: PMC4855979
- DOI: 10.1186/s13059-016-0935-y
An evaluation of methods correcting for cell-type heterogeneity in DNA methylation studies
Abstract
Background: Many different methods exist to adjust for variability in cell-type mixture proportions when analyzing DNA methylation studies. Here we present the result of an extensive simulation study, built on cell-separated DNA methylation profiles from Illumina Infinium 450K methylation data, to compare the performance of eight methods including the most commonly used approaches.
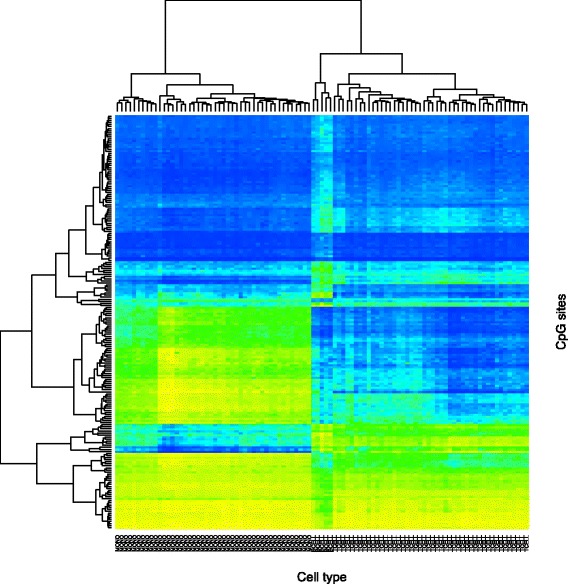
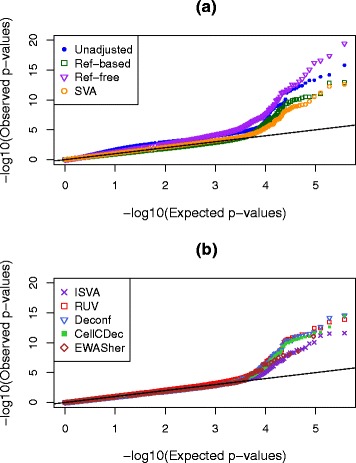
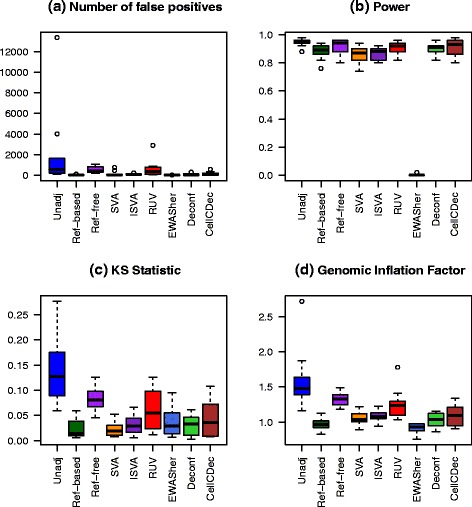
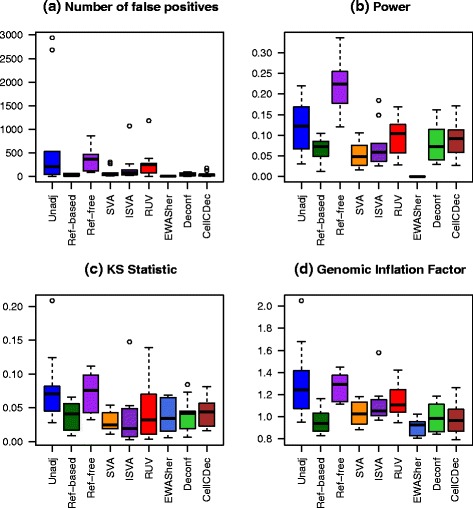
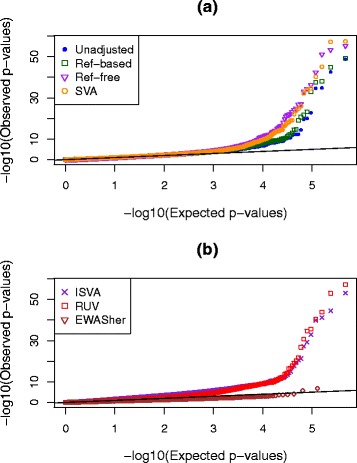
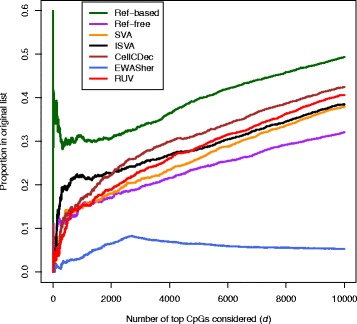
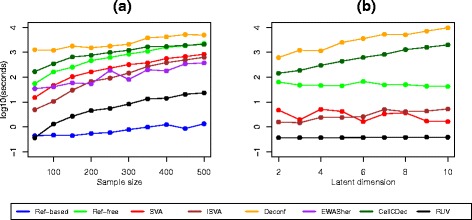
Results: We designed a rich multi-layered simulation containing a set of probes with true associations with either binary or continuous phenotypes, confounding by cell type, variability in means and standard deviations for population parameters, additional variability at the level of an individual cell-type-specific sample, and variability in the mixture proportions across samples. Performance varied quite substantially across methods and simulations. In particular, the number of false positives was sometimes unrealistically high, indicating limited ability to discriminate the true signals from those appearing significant through confounding. Methods that filtered probes had consequently poor power. QQ plots of p values across all tested probes showed that adjustments did not always improve the distribution. The same methods were used to examine associations between smoking and methylation data from a case-control study of colorectal cancer, and we also explored the effect of cell-type adjustments on associations between rheumatoid arthritis cases and controls.
Conclusions: We recommend surrogate variable analysis for cell-type mixture adjustment since performance was stable under all our simulated scenarios.
Keywords: Cell-type mixture; DNA methylation; Deconvolution; Matrix decomposition.
Figures
Comment in
-
Correcting for cell-type effects in DNA methylation studies: reference-based method outperforms latent variable approaches in empirical studies.Genome Biol. 2017 Jan 30;18(1):24. doi: 10.1186/s13059-017-1148-8. Genome Biol. 2017. PMID: 28137292 Free PMC article.
Similar articles
-
The correlation of methylation levels measured using Illumina 450K and EPIC BeadChips in blood samples.Epigenomics. 2017 Nov;9(11):1363-1371. doi: 10.2217/epi-2017-0078. Epub 2017 Aug 15. Epigenomics. 2017. PMID: 28809127 Free PMC article.
-
Improving cell mixture deconvolution by identifying optimal DNA methylation libraries (IDOL).BMC Bioinformatics. 2016 Mar 8;17:120. doi: 10.1186/s12859-016-0943-7. BMC Bioinformatics. 2016. PMID: 26956433 Free PMC article.
-
Guidelines for cell-type heterogeneity quantification based on a comparative analysis of reference-free DNA methylation deconvolution software.BMC Bioinformatics. 2020 Jan 13;21(1):16. doi: 10.1186/s12859-019-3307-2. BMC Bioinformatics. 2020. PMID: 31931698 Free PMC article.
-
Cell-type deconvolution from DNA methylation: a review of recent applications.Hum Mol Genet. 2017 Oct 1;26(R2):R216-R224. doi: 10.1093/hmg/ddx275. Hum Mol Genet. 2017. PMID: 28977446 Free PMC article. Review.
-
Subgroup analyses in randomised controlled trials: quantifying the risks of false-positives and false-negatives.Health Technol Assess. 2001;5(33):1-56. doi: 10.3310/hta5330. Health Technol Assess. 2001. PMID: 11701102 Review.
Cited by
-
Systematic evaluation and validation of reference and library selection methods for deconvolution of cord blood DNA methylation data.Clin Epigenetics. 2019 Aug 27;11(1):125. doi: 10.1186/s13148-019-0717-y. Clin Epigenetics. 2019. PMID: 31455416 Free PMC article.
-
Sparse principal component analysis based on genome network for correcting cell type heterogeneity in epigenome-wide association studies.Med Biol Eng Comput. 2022 Sep;60(9):2601-2618. doi: 10.1007/s11517-022-02599-9. Epub 2022 Jul 4. Med Biol Eng Comput. 2022. PMID: 35789457
-
Detection of cell-type-specific differentially methylated regions in epigenome-wide association studies.Bioinformatics. 2025 Jul 1;41(Supplement_1):i502-i512. doi: 10.1093/bioinformatics/btaf243. Bioinformatics. 2025. PMID: 40662795 Free PMC article.
-
A Data-Driven Epigenetic Characterization of Morning Fatigue Severity in Oncology Patients Receiving Chemotherapy: Associations With Epigenetic Age Acceleration, Blood Cell Types, and Expression-Associated Methylation.Cancer Med. 2025 Aug;14(15):e71067. doi: 10.1002/cam4.71067. Cancer Med. 2025. PMID: 40709591 Free PMC article.
-
Whole genome methylation and transcriptome analyses to identify risk for cerebral palsy (CP) in extremely low gestational age neonates (ELGAN).Sci Rep. 2021 Mar 5;11(1):5305. doi: 10.1038/s41598-021-84214-9. Sci Rep. 2021. PMID: 33674671 Free PMC article. Clinical Trial.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
