

Protein Scaffolds Control Localized Protein Kinase Cζ Activity
- PMID: 27143478
- PMCID: PMC4919463
- DOI: 10.1074/jbc.M116.729483
Protein Scaffolds Control Localized Protein Kinase Cζ Activity
Abstract
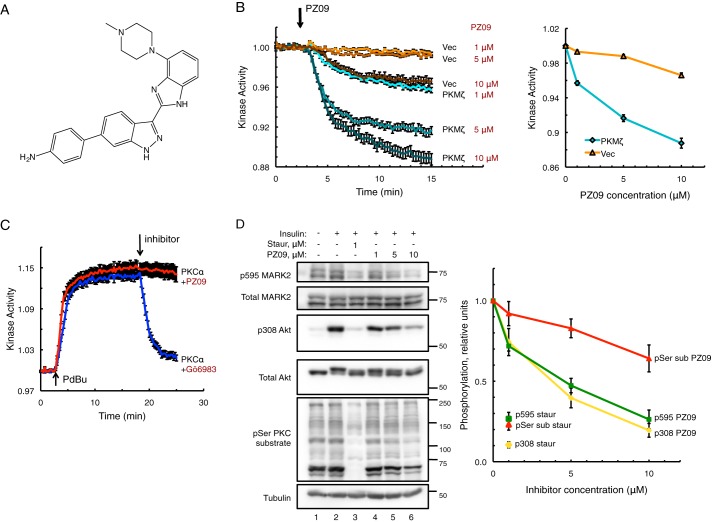
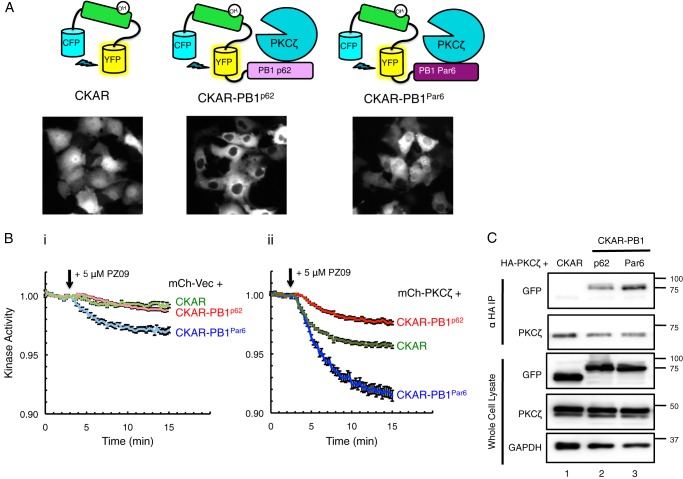
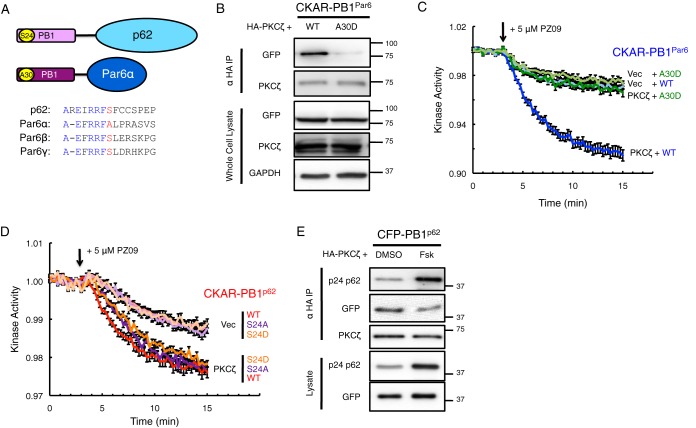
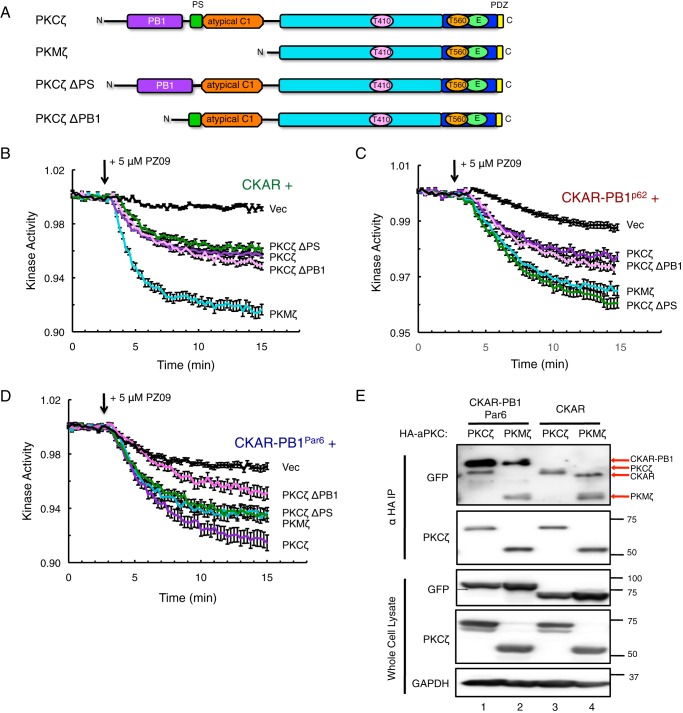
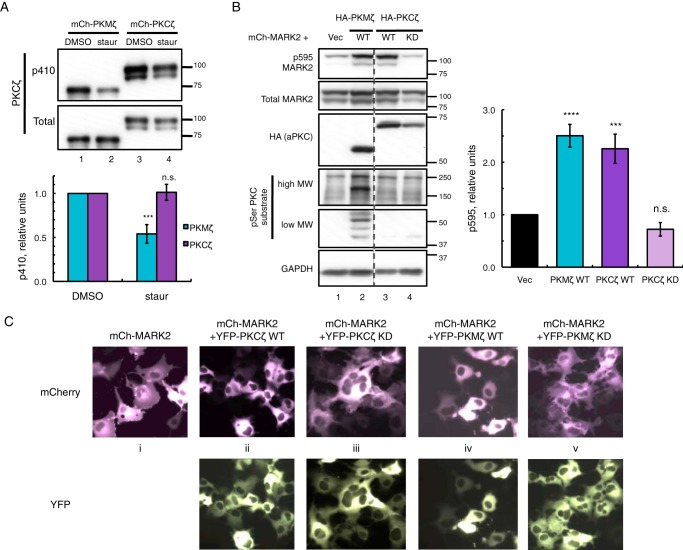
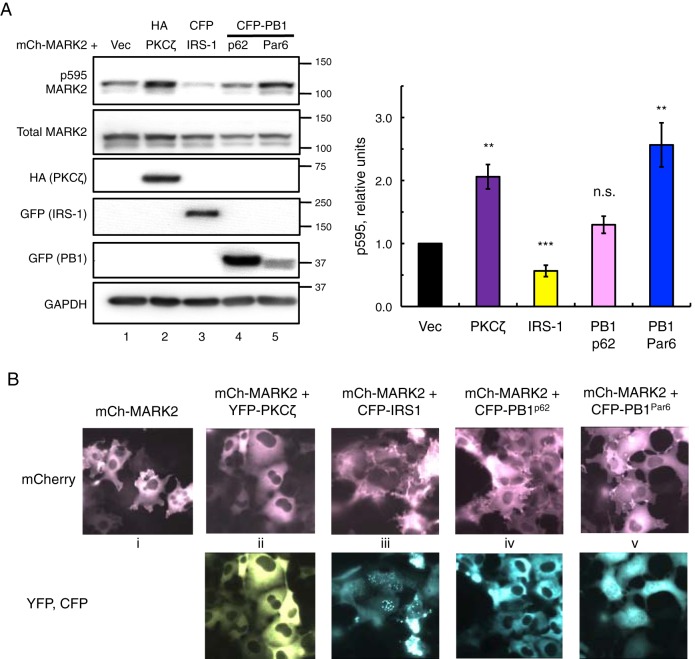
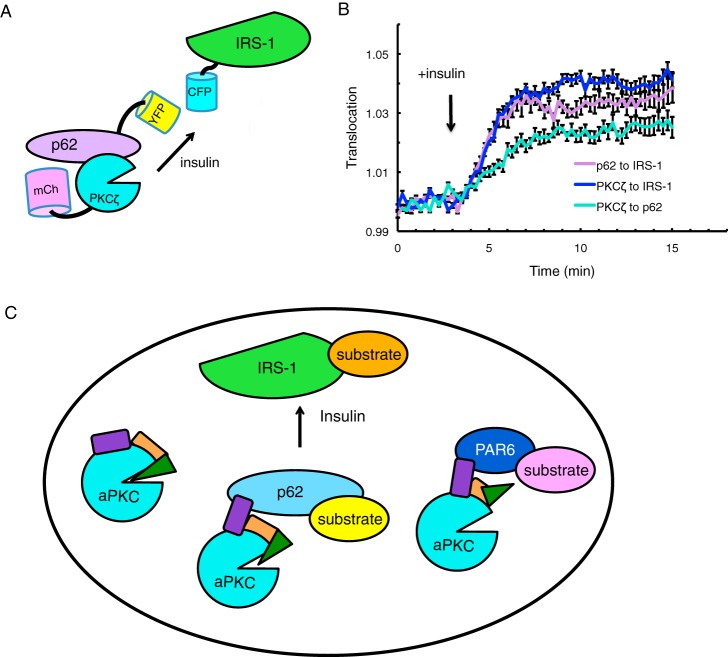
Atypical protein kinase C (aPKC) isozymes modulate insulin signaling and cell polarity, but how their activity is controlled in cells is not well understood. These enzymes are constitutively phosphorylated, insensitive to second messengers, and have relatively low activity. Here we show that protein scaffolds not only localize but also differentially control the catalytic activity of the aPKC PKCζ, thus promoting activity toward localized substrates and restricting activity toward global substrates. Using cellular substrate readouts and scaffolded activity reporters in live cell imaging, we show that PKCζ has highly localized and differentially controlled activity on the scaffolds p62 and Par6. Both scaffolds tether aPKC in an active conformation as assessed through pharmacological inhibition of basal activity, monitored using a genetically encoded reporter for PKC activity. However, binding to Par6 is of higher affinity and is more effective in locking PKCζ in an active conformation. FRET-based translocation assays reveal that insulin promotes the association of both p62 and aPKC with the insulin-regulated scaffold IRS-1. Using the aPKC substrate MARK2 as another readout for activity, we show that overexpression of IRS-1 reduces the phosphorylation of MARK2 and enhances its plasma membrane localization, indicating sequestration of aPKC by IRS-1 away from MARK2. These results are consistent with scaffolds serving as allosteric activators of aPKCs, tethering them in an active conformation near specific substrates. Thus, signaling of these intrinsically low activity kinases is kept at a minimum in the absence of scaffolding interactions, which position the enzymes for stoichiometric phosphorylation of substrates co-localized on the same protein scaffold.
Keywords: MARK2; Par6; autoinhibition; insulin; insulin receptor substrate 1 (IRS-1); p62 (sequestosome 1(SQSTM1)); protein kinase C (PKC); scaffold protein.
© 2016 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
Similar articles
-
Zeta Inhibitory Peptide Disrupts Electrostatic Interactions That Maintain Atypical Protein Kinase C in Its Active Conformation on the Scaffold p62.J Biol Chem. 2015 Sep 4;290(36):21845-56. doi: 10.1074/jbc.M115.676221. Epub 2015 Jul 17. J Biol Chem. 2015. PMID: 26187466 Free PMC article.
-
Sequestosome 1/p62, a scaffolding protein, is a newly identified partner of IRS-1 protein.J Biol Chem. 2012 Aug 24;287(35):29672-8. doi: 10.1074/jbc.M111.322404. Epub 2012 Jul 3. J Biol Chem. 2012. PMID: 22761437 Free PMC article.
-
Protein kinase Cζ exhibits constitutive phosphorylation and phosphatidylinositol-3,4,5-triphosphate-independent regulation.Biochem J. 2016 Feb 15;473(4):509-23. doi: 10.1042/BJ20151013. Epub 2015 Dec 3. Biochem J. 2016. PMID: 26635352 Free PMC article.
-
Atypical protein kinase C in cardiometabolic abnormalities.Curr Opin Lipidol. 2012 Jun;23(3):175-181. doi: 10.1097/MOL.0b013e328352c4c7. Curr Opin Lipidol. 2012. PMID: 22449812 Free PMC article. Review.
-
Of the atypical PKCs, Par-4 and p62: recent understandings of the biology and pathology of a PB1-dominated complex.Cell Death Differ. 2009 Nov;16(11):1426-37. doi: 10.1038/cdd.2009.119. Epub 2009 Aug 28. Cell Death Differ. 2009. PMID: 19713972 Free PMC article. Review.
Cited by
-
Activation of atypical protein kinase C by sphingosine 1-phosphate revealed by an aPKC-specific activity reporter.Sci Signal. 2019 Jan 1;12(562):eaat6662. doi: 10.1126/scisignal.aat6662. Sci Signal. 2019. PMID: 30600259 Free PMC article.
-
Role of Protein Kinase C in Immune Cell Activation and Its Implication Chemical-Induced Immunotoxicity.Adv Exp Med Biol. 2021;1275:151-163. doi: 10.1007/978-3-030-49844-3_6. Adv Exp Med Biol. 2021. PMID: 33539015
-
Sensitive fluorescent biosensor reveals differential subcellular regulation of PKC.Nat Chem Biol. 2025 Apr;21(4):501-511. doi: 10.1038/s41589-024-01758-3. Epub 2024 Oct 11. Nat Chem Biol. 2025. PMID: 39394268
-
A Multimodel Study of the Role of Novel PKC Isoforms in the DNA Integrity Checkpoint.Int J Mol Sci. 2023 Oct 31;24(21):15796. doi: 10.3390/ijms242115796. Int J Mol Sci. 2023. PMID: 37958781 Free PMC article.
-
Protein kinase C mechanisms that contribute to cardiac remodelling.Clin Sci (Lond). 2016 Sep 1;130(17):1499-510. doi: 10.1042/CS20160036. Clin Sci (Lond). 2016. PMID: 27433023 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
