

Culturing of 'unculturable' human microbiota reveals novel taxa and extensive sporulation
- PMID: 27144353
- PMCID: PMC4890681
- DOI: 10.1038/nature17645
Culturing of 'unculturable' human microbiota reveals novel taxa and extensive sporulation
Abstract
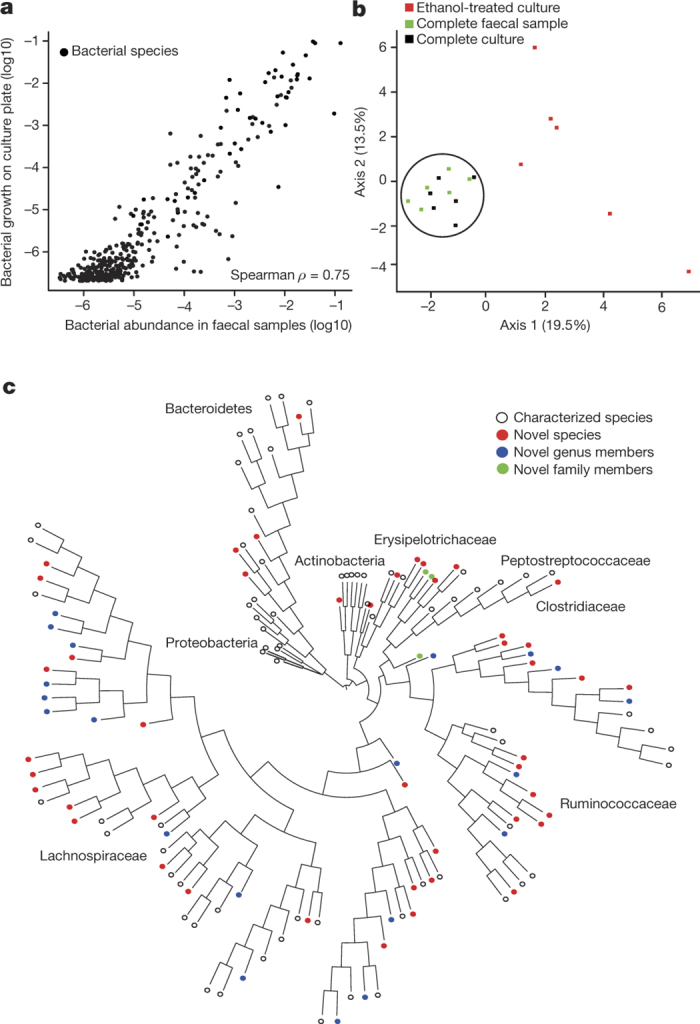
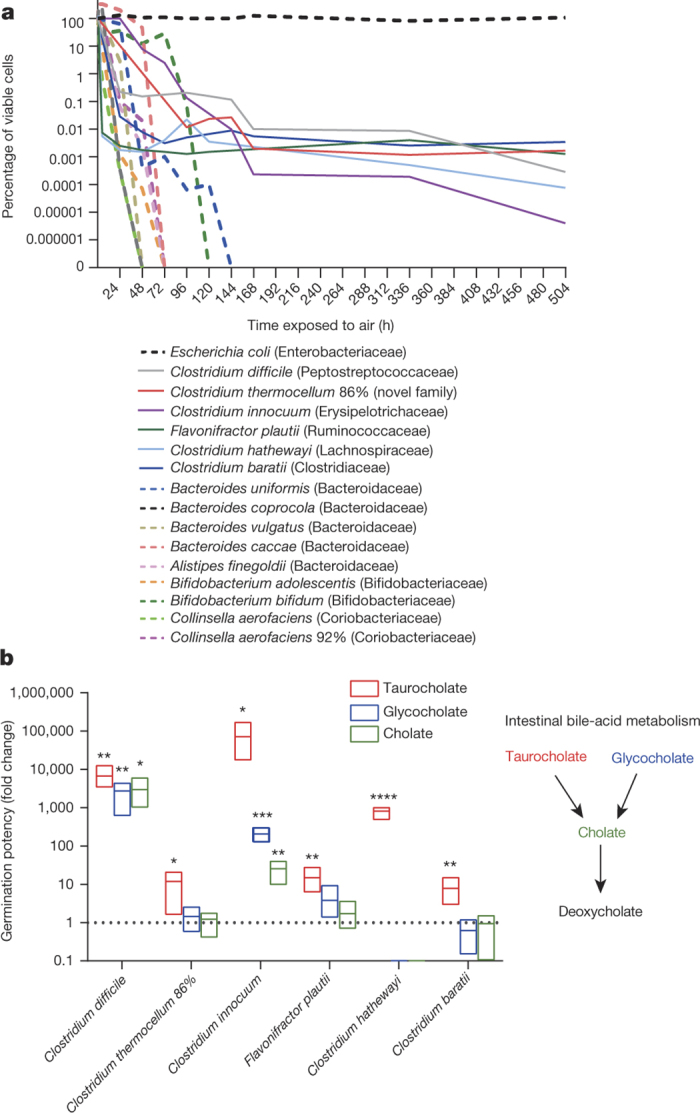
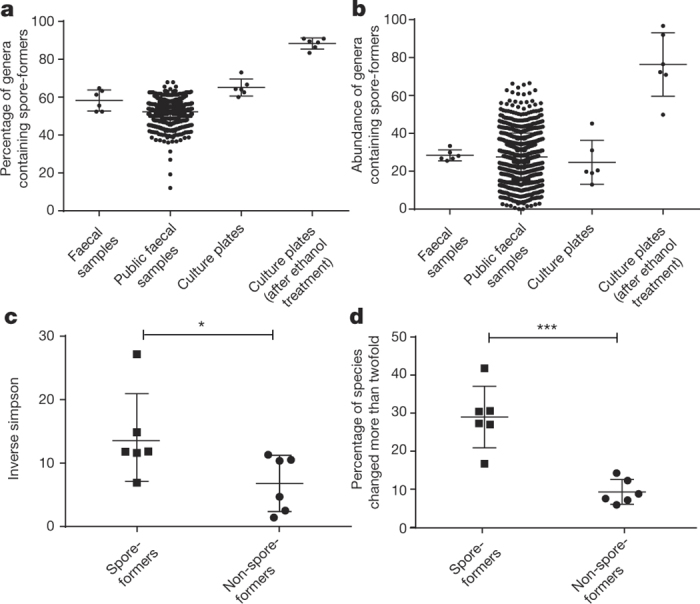
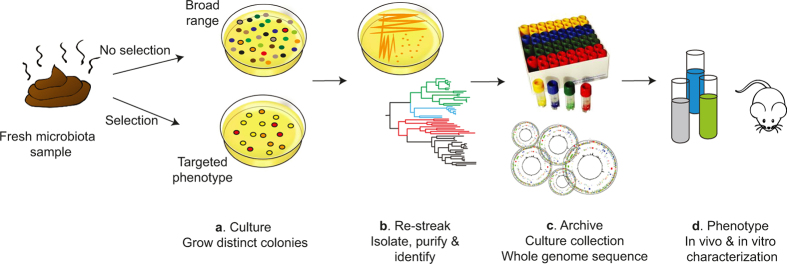
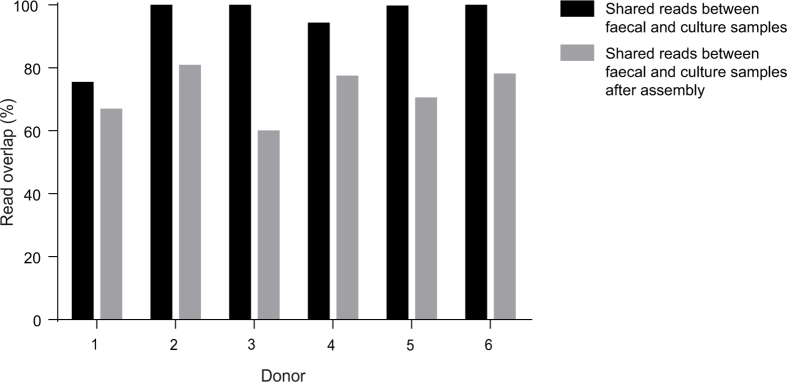
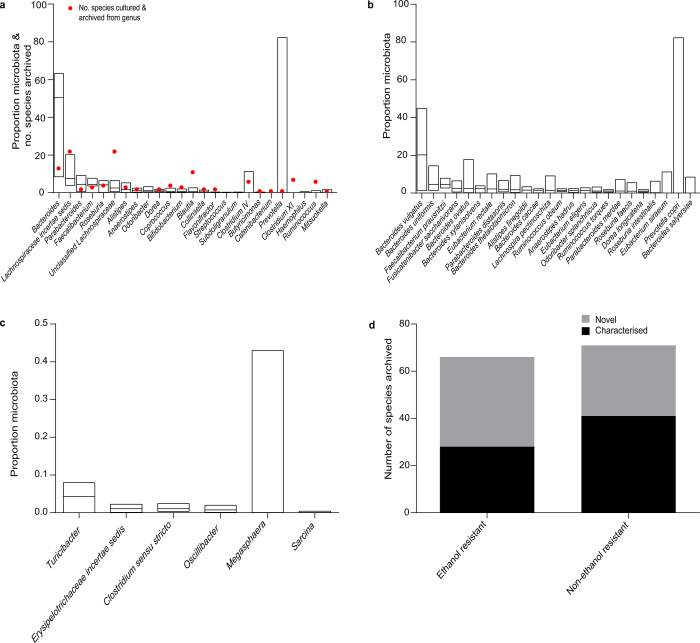
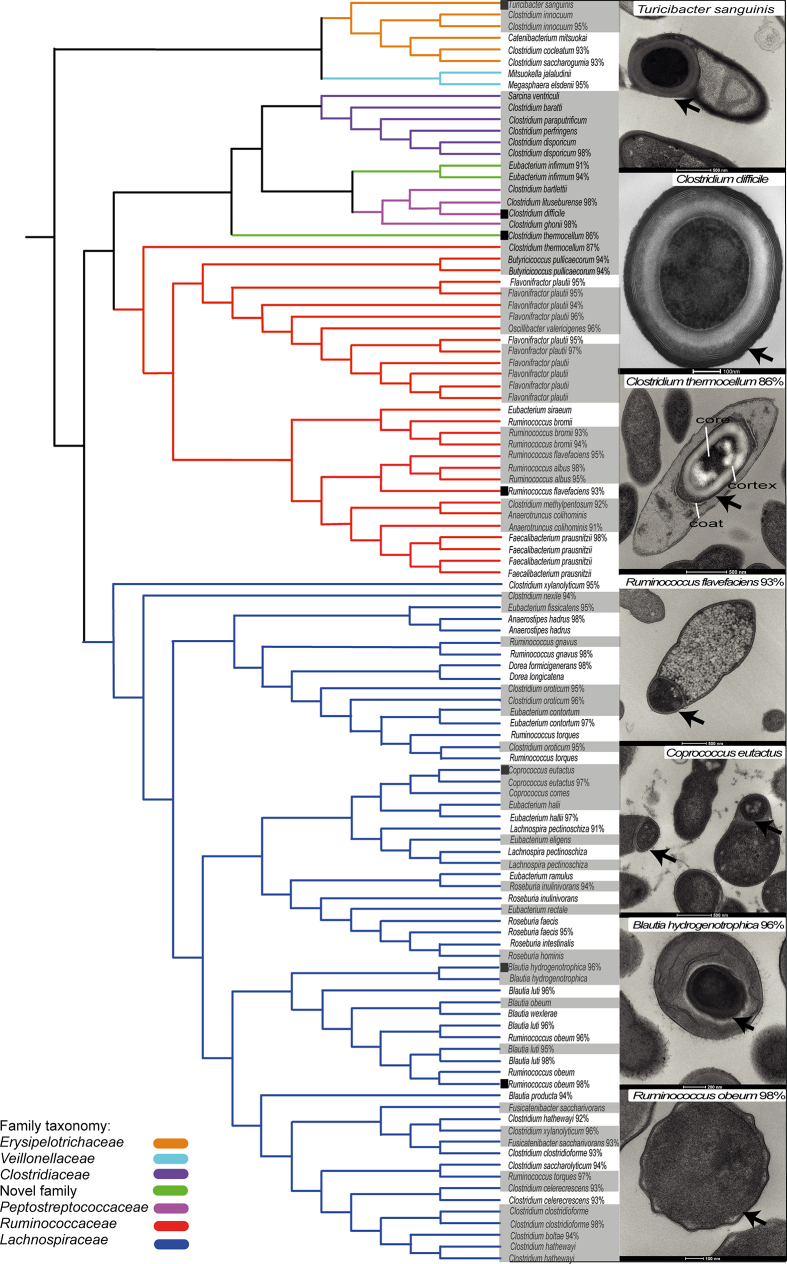
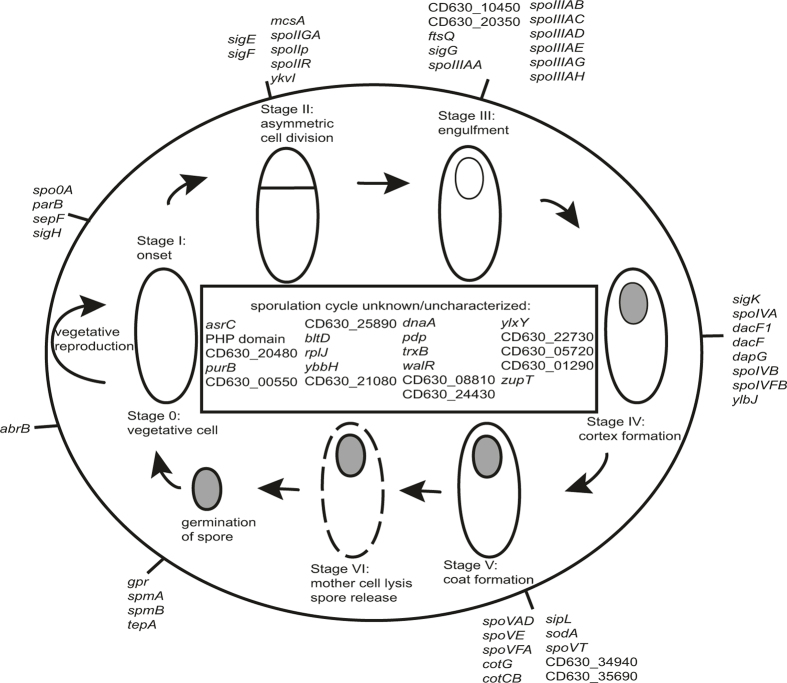
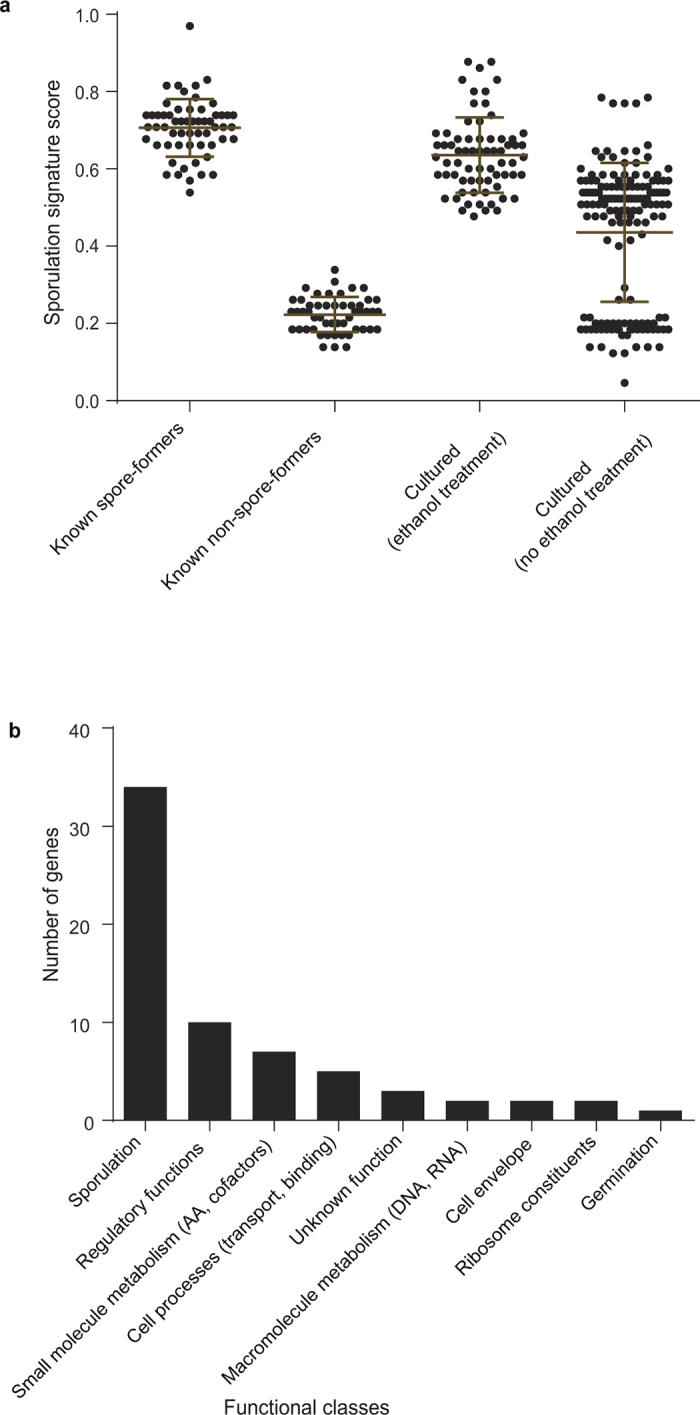
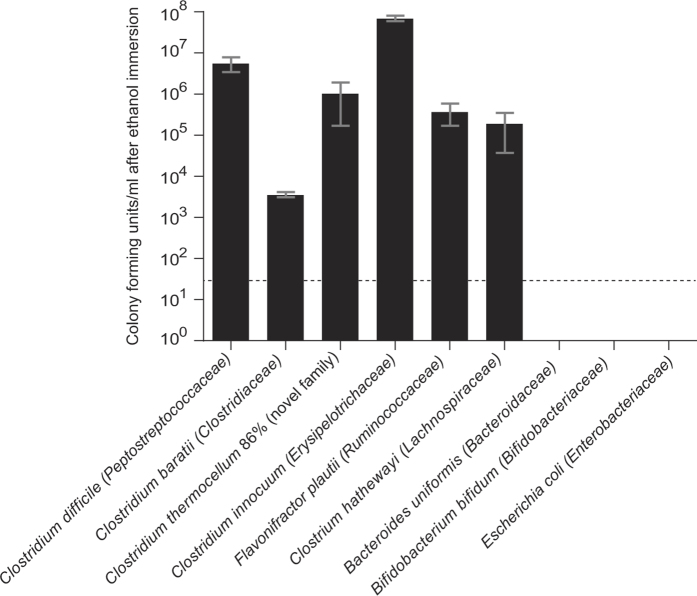
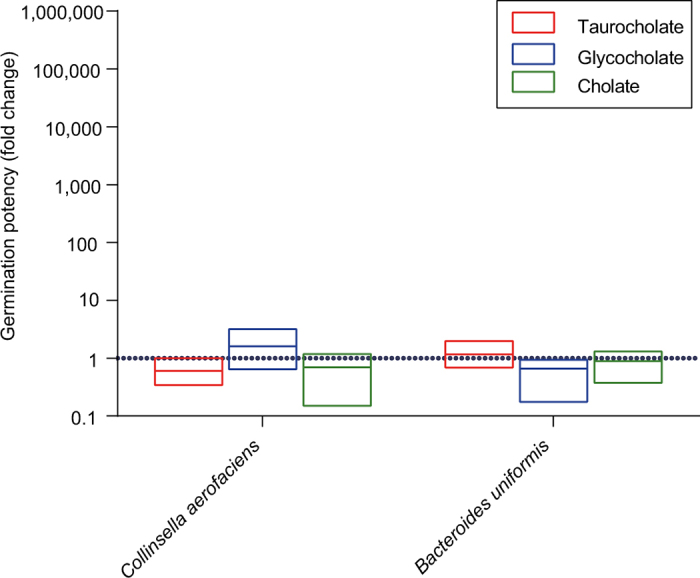
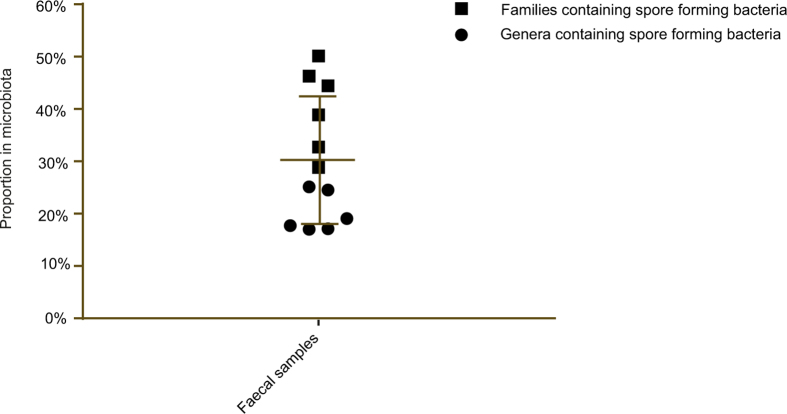
Our intestinal microbiota harbours a diverse bacterial community required for our health, sustenance and wellbeing. Intestinal colonization begins at birth and climaxes with the acquisition of two dominant groups of strict anaerobic bacteria belonging to the Firmicutes and Bacteroidetes phyla. Culture-independent, genomic approaches have transformed our understanding of the role of the human microbiome in health and many diseases. However, owing to the prevailing perception that our indigenous bacteria are largely recalcitrant to culture, many of their functions and phenotypes remain unknown. Here we describe a novel workflow based on targeted phenotypic culturing linked to large-scale whole-genome sequencing, phylogenetic analysis and computational modelling that demonstrates that a substantial proportion of the intestinal bacteria are culturable. Applying this approach to healthy individuals, we isolated 137 bacterial species from characterized and candidate novel families, genera and species that were archived as pure cultures. Whole-genome and metagenomic sequencing, combined with computational and phenotypic analysis, suggests that at least 50-60% of the bacterial genera from the intestinal microbiota of a healthy individual produce resilient spores, specialized for host-to-host transmission. Our approach unlocks the human intestinal microbiota for phenotypic analysis and reveals how a marked proportion of oxygen-sensitive intestinal bacteria can be transmitted between individuals, affecting microbiota heritability.
Conflict of interest statement
Competing Interests. The authors have filed a patent application related to the described work.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
