

The role of ER stress in lipid metabolism and lipotoxicity
- PMID: 27146479
- PMCID: PMC4959874
- DOI: 10.1194/jlr.R067595
The role of ER stress in lipid metabolism and lipotoxicity
Abstract
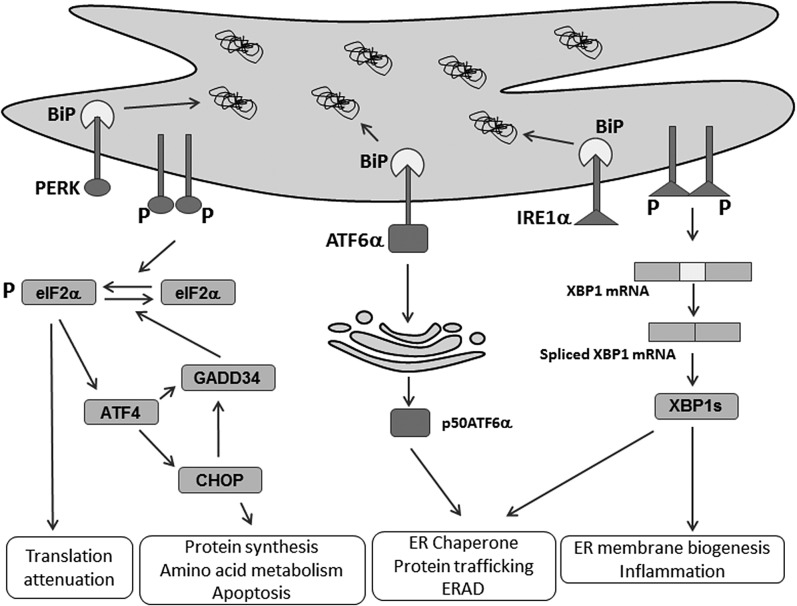
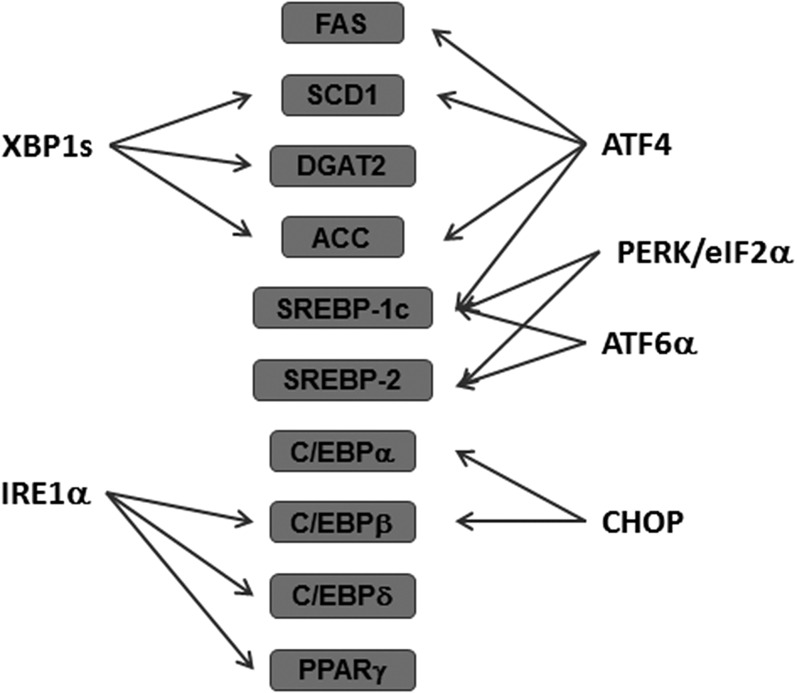
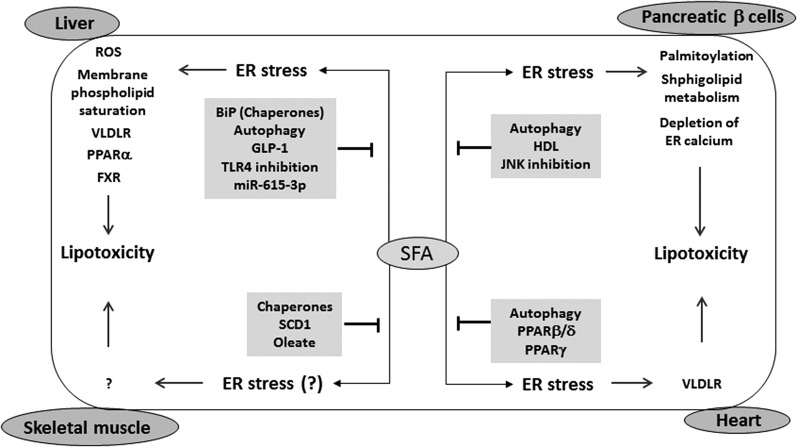
The endoplasmic reticulum (ER) is a cellular organelle important for regulating calcium homeostasis, lipid metabolism, protein synthesis, and posttranslational modification and trafficking. Numerous environmental, physiological, and pathological insults disturb ER homeostasis, referred to as ER stress, in which a collection of conserved intracellular signaling pathways, termed the unfolded protein response (UPR), are activated to maintain ER function for cell survival. However, excessive and/or prolonged UPR activation leads to initiation of self-destruction through apoptosis. Excessive accumulation of lipids and their intermediate products causes metabolic abnormalities and cell death, called lipotoxicity, in peripheral organs, including the pancreatic islets, liver, muscle, and heart. Because accumulating evidence links chronic ER stress and defects in UPR signaling to lipotoxicity in peripheral tissues, understanding the role of ER stress in cell physiology is a topic under intense investigation. In this review, we highlight recent findings that link ER stress and UPR signaling to the pathogenesis of peripheral organs due to lipotoxicity.
Keywords: cell signaling; diabetes; endoplasmic reticulum; fatty acid; lipids.
Copyright © 2016 by the American Society for Biochemistry and Molecular Biology, Inc.
Figures
Similar articles
-
Endoplasmic reticulum stress and lipids in health and diseases.Prog Lipid Res. 2023 Jan;89:101198. doi: 10.1016/j.plipres.2022.101198. Epub 2022 Nov 13. Prog Lipid Res. 2023. PMID: 36379317 Review.
-
Emerging roles of ER stress and unfolded protein response pathways in skeletal muscle health and disease.J Cell Physiol. 2018 Jan;233(1):67-78. doi: 10.1002/jcp.25852. Epub 2017 May 16. J Cell Physiol. 2018. PMID: 28177127 Free PMC article. Review.
-
The essential functions of endoplasmic reticulum chaperones in hepatic lipid metabolism.Dig Liver Dis. 2016 Jul;48(7):709-16. doi: 10.1016/j.dld.2016.03.016. Epub 2016 Mar 31. Dig Liver Dis. 2016. PMID: 27133206 Review.
-
Lipotoxicity, ER Stress, and Cardiovascular Disease: Current Understanding and Future Directions.Cardiovasc Hematol Agents Med Chem. 2024;22(3):319-335. doi: 10.2174/0118715257262366230928051902. Cardiovasc Hematol Agents Med Chem. 2024. PMID: 37859305 Review.
-
The unfolded protein response and hepatic lipid metabolism in non alcoholic fatty liver disease.Pharmacol Ther. 2019 Nov;203:107401. doi: 10.1016/j.pharmthera.2019.107401. Epub 2019 Aug 13. Pharmacol Ther. 2019. PMID: 31419516 Free PMC article. Review.
Cited by
-
A forebrain-hypothalamic ER stress driven circuit mediates hepatic steatosis during obesity.Mol Metab. 2024 Jan;79:101858. doi: 10.1016/j.molmet.2023.101858. Epub 2023 Dec 21. Mol Metab. 2024. PMID: 38141847 Free PMC article.
-
Uremic Sarcopenia: Clinical Evidence and Basic Experimental Approach.Nutrients. 2020 Jun 18;12(6):1814. doi: 10.3390/nu12061814. Nutrients. 2020. PMID: 32570738 Free PMC article. Review.
-
Endoplasmic Reticulum Stress in Macrophages: The Vicious Circle of Lipid Accumulation and Pro-Inflammatory Response.Biomedicines. 2020 Jul 13;8(7):210. doi: 10.3390/biomedicines8070210. Biomedicines. 2020. PMID: 32668733 Free PMC article. Review.
-
Increasing Fatty Acid Oxidation Prevents High-Fat Diet-Induced Cardiomyopathy Through Regulating Parkin-Mediated Mitophagy.Circulation. 2020 Sep 8;142(10):983-997. doi: 10.1161/CIRCULATIONAHA.119.043319. Epub 2020 Jun 29. Circulation. 2020. PMID: 32597196 Free PMC article.
-
Current Understanding on the Role of Lipids in Macrophages and Associated Diseases.Int J Mol Sci. 2022 Dec 29;24(1):589. doi: 10.3390/ijms24010589. Int J Mol Sci. 2022. PMID: 36614031 Free PMC article. Review.
References
-
- Clapham D. E. 2007. Calcium signaling. Cell. 131: 1047–1058. - PubMed
-
- Moore L., Chen T., Knapp H. R. Jr., and Landon E. J.. 1975. Energy-dependent calcium sequestration activity in rat liver microsomes. J. Biol. Chem. 250: 4562–4568. - PubMed
-
- Michalak M., Robert Parker J. M., and Opas M.. 2002. Ca2+ signaling and calcium binding chaperones of the endoplasmic reticulum. Cell Calcium. 32: 269–278. - PubMed
-
- Schröder M., and Kaufman R. J.. 2005. The mammalian unfolded protein response. Annu. Rev. Biochem. 74: 739–789. - PubMed
-
- Wang M., and Kaufman R. J.. 2016. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature. 529: 326–335. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
