

The transcription factor NeuroD2 coordinates synaptic innervation and cell intrinsic properties to control excitability of cortical pyramidal neurons
- PMID: 27146976
- PMCID: PMC4929321
- DOI: 10.1113/JP271953
The transcription factor NeuroD2 coordinates synaptic innervation and cell intrinsic properties to control excitability of cortical pyramidal neurons
Abstract
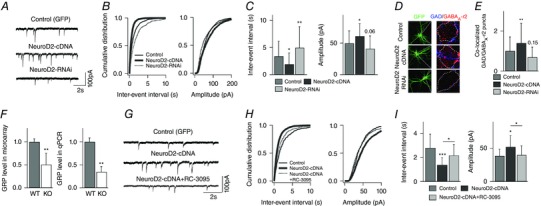
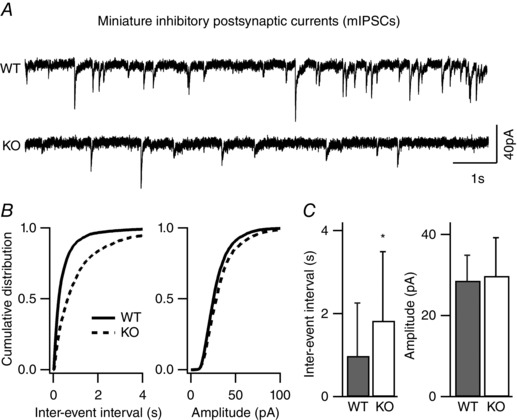
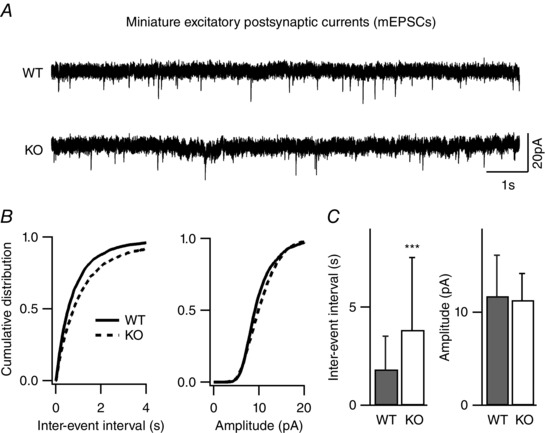
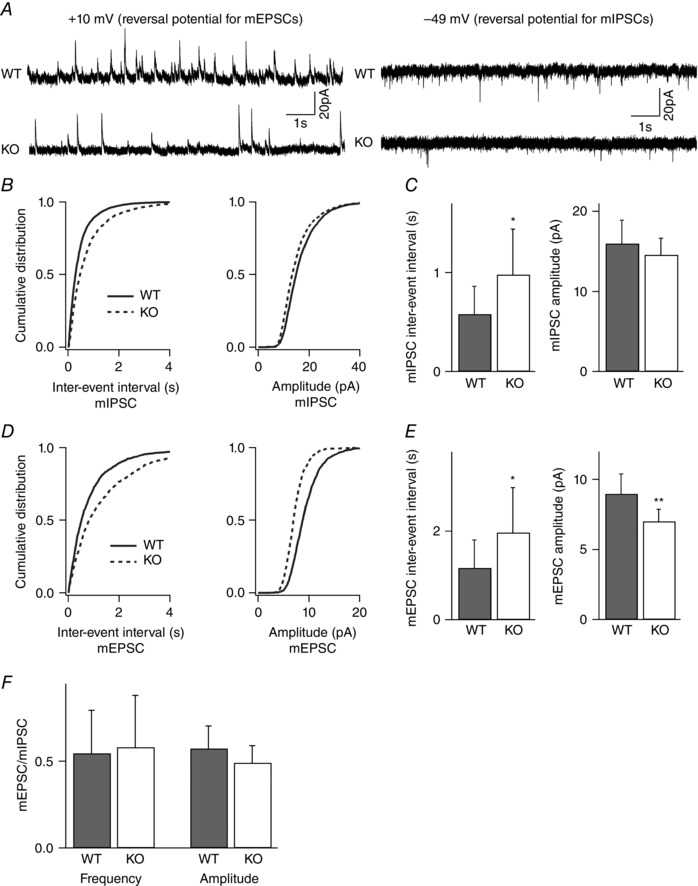
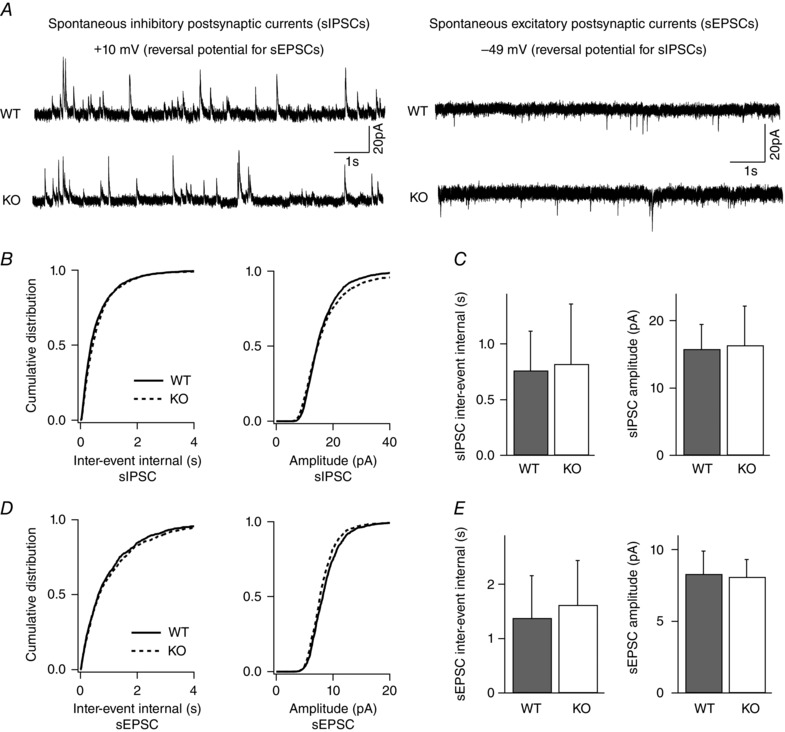
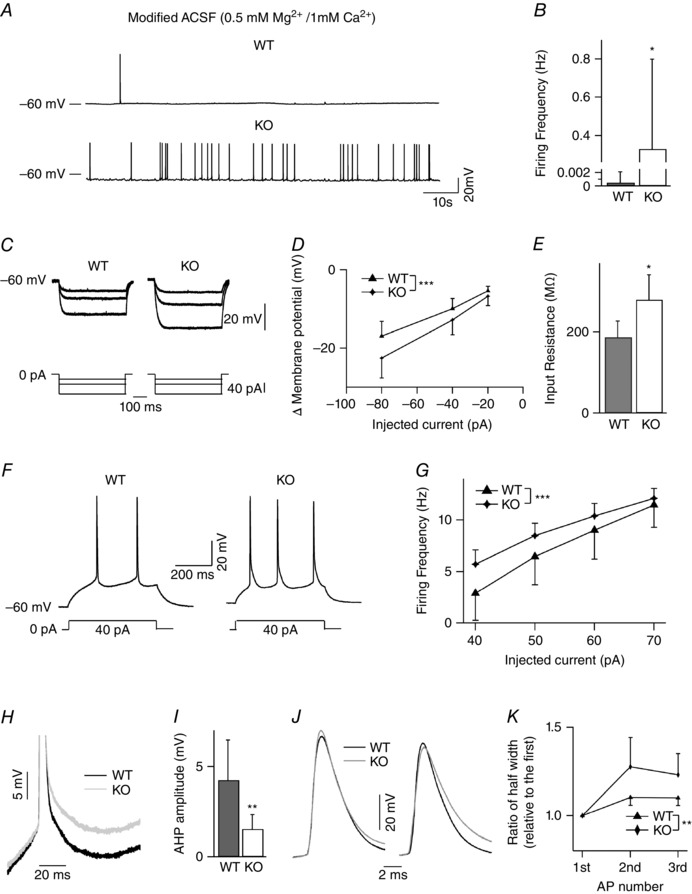
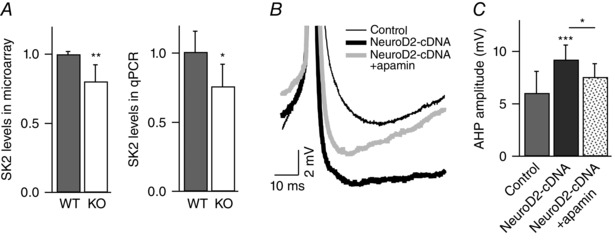
Key points: Synaptic excitation and inhibition must be properly balanced in individual neurons and neuronal networks to allow proper brain function. Disrupting this balance may lead to autism spectral disorders and epilepsy. We show the basic helix-loop-helix transcription factor NeuroD2 promotes inhibitory synaptic drive but also decreases cell-intrinsic neuronal excitability of cortical pyramidal neurons both in vitro and in vivo. We identify two genes potentially downstream of NeuroD2-mediated transcription that regulate these parameters: gastrin-releasing peptide and the small conductance, calcium-activated potassium channel, SK2. Our results reveal an important function for NeuroD2 in balancing synaptic neurotransmission and intrinsic excitability. Our results offer insight into how synaptic innervation and intrinsic excitability are coordinated during cortical development.
Abstract: Synaptic excitation and inhibition must be properly balanced in individual neurons and neuronal networks for proper brain function. Disruption of this balance during development may lead to autism spectral disorders and epilepsy. Synaptic excitation is counterbalanced by synaptic inhibition but also by attenuation of cell-intrinsic neuronal excitability. To maintain proper excitation levels during development, neurons must sense activity over time and regulate the expression of genes that control these parameters. While this is a critical process, little is known about the transcription factors involved in coordinating gene expression to control excitatory/inhibitory synaptic balance. We show here that the basic helix-loop-helix transcription factor NeuroD2 promotes inhibitory synaptic drive but also decreases cell-intrinsic neuronal excitability of cortical pyramidal neurons both in vitro and in vivo as shown by ex vivo analysis of a NeuroD2 knockout mouse. Using microarray analysis and comparing wild-type and NeuroD2 knockout cortical networks, we identified two potential gene targets of NeuroD2 that contribute to these processes: gastrin-releasing peptide (GRP) and the small conductance, calcium-activated potassium channel, SK2. We found that the GRP receptor antagonist RC-3059 and the SK2 specific blocker apamin partially reversed the effects of increased NeuroD2 expression on inhibitory synaptic drive and action potential repolarization, respectively. Our results reveal an important function for NeuroD2 in balancing synaptic neurotransmission and intrinsic excitability and offer insight into how these processes are coordinated during cortical development.
© 2016 The Authors. The Journal of Physiology © 2016 The Physiological Society.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases