

Prevention effect of rare ginsenosides against stress-hormone induced MTOC amplification
- PMID: 27147573
- PMCID: PMC5085216
- DOI: 10.18632/oncotarget.9059
Prevention effect of rare ginsenosides against stress-hormone induced MTOC amplification
Abstract
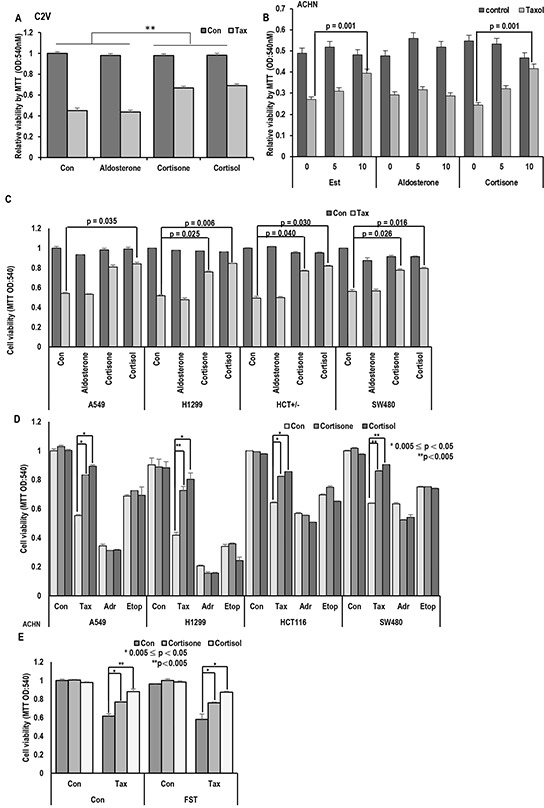
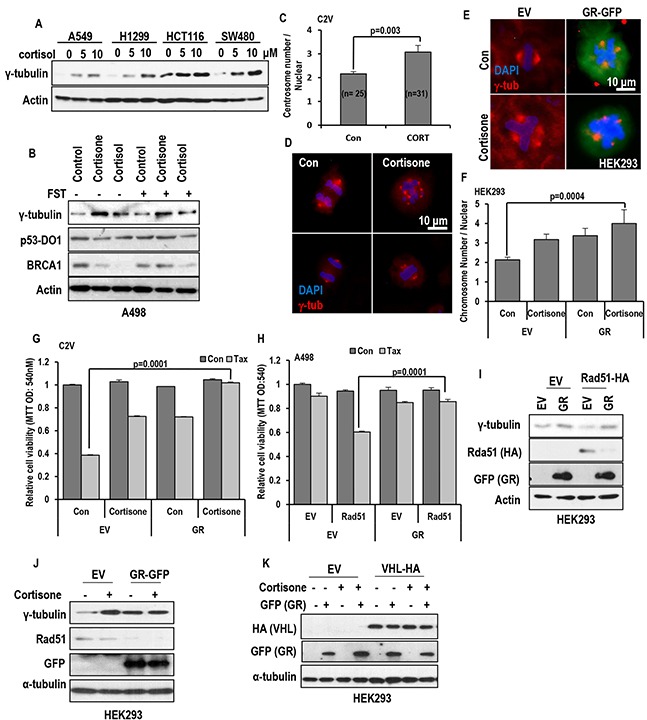
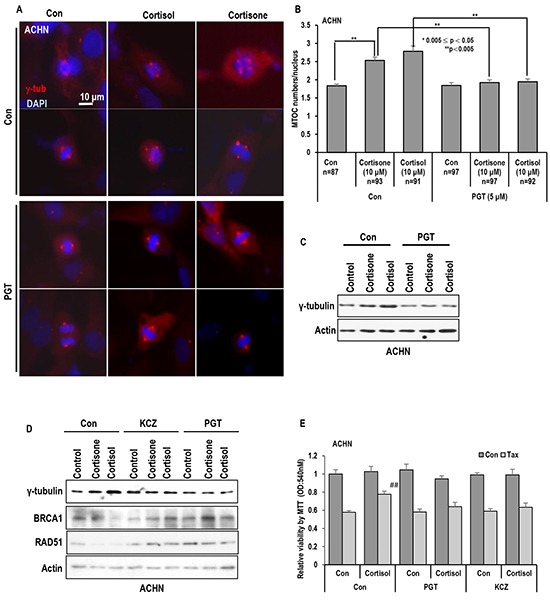
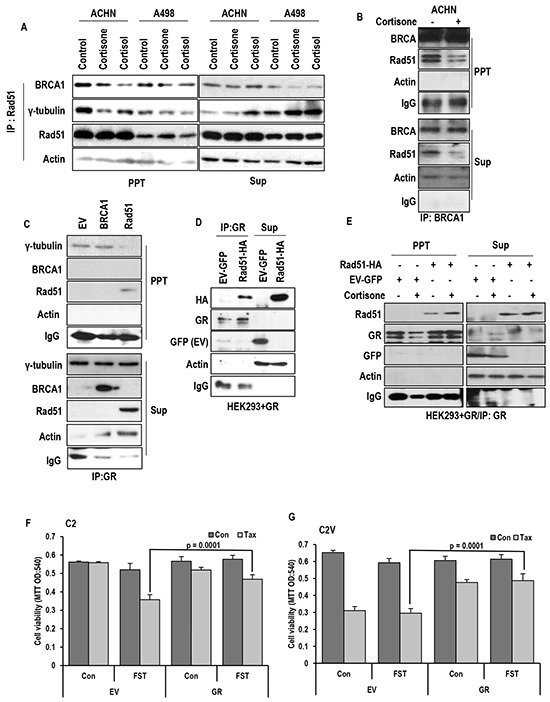
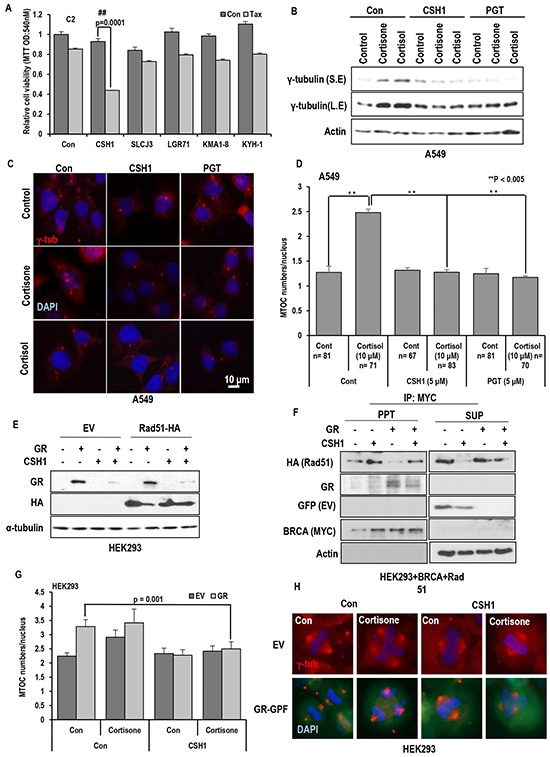
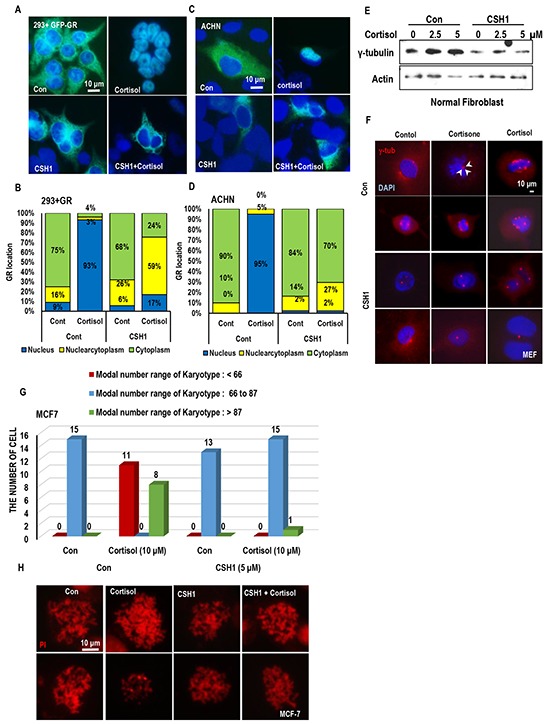
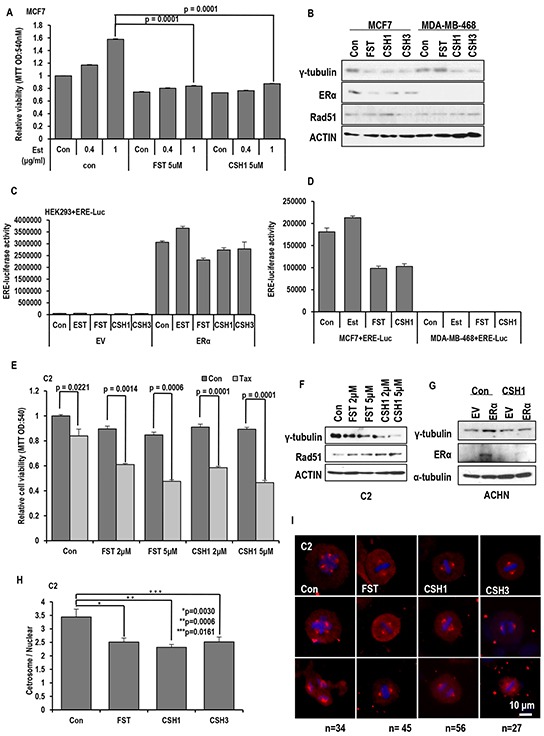
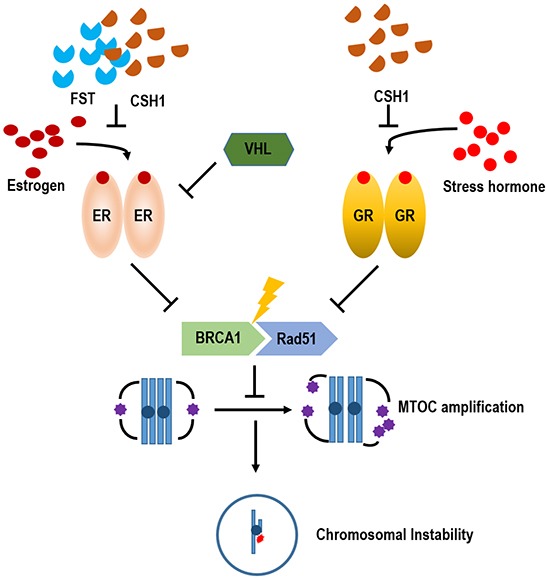
Stress has been suggested as one of important cause of human cancer without molecular biological evidence. Thus, we test the effect of stress-related hormones on cell viability and mitotic fidelity. Similarly to estrogen, stress hormone cortisol and its relative cortisone increase microtubule organizing center (MTOC) number through elevated expression of γ-tubulin and provide the Taxol resistance to human cancer cell lines. However, these effects are achieved by glucocorticoid hormone receptor (GR) but not by estrogen receptor (ER). Since ginsenosides possess steroid-like structure, we hypothesized that it would block the stress or estrogen-induced MTOC amplification and Taxol resistance. Among tested chemicals, rare ginsenoside, CSH1 (Rg6) shows obvious effect on inhibition of MTOC amplification, γ-tubulin induction and Taxol resistance. Comparing to Fulvestant (FST), ER-α specific inhibitor, this chemical can block the cortisol/cortisone-induced MTOC deregulation as well as ER-α signaling. Our results suggest that stress hormone induced tumorigenesis would be achieved by MTOC amplification, and CSH1 would be useful for prevention of stress-hormone or steroid hormone-induced chromosomal instability.
Keywords: MTOC; cancer prevention; stress hormone.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
Elevated estrogen receptor-α in VHL-deficient condition induces microtubule organizing center amplification via disruption of BRCA1/Rad51 interaction.Neoplasia. 2014 Dec;16(12):1070-81. doi: 10.1016/j.neo.2014.09.013. Neoplasia. 2014. PMID: 25499220 Free PMC article.
-
Microtubule-organizing center-mediated nuclear polarity in various normal and neoplastic human tissues.Virchows Arch. 2015 Jun;466(6):625-35. doi: 10.1007/s00428-015-1744-5. Epub 2015 Mar 6. Virchows Arch. 2015. PMID: 25742907
-
Effects of the herbicide isopropyl-N-phenyl carbamate on microtubules and MTOCs in lines of Nicotiana sylvestris resistant and sensitive to its action.Cell Biol Int. 2008 Jun;32(6):623-9. doi: 10.1016/j.cellbi.2008.01.012. Epub 2008 Jan 25. Cell Biol Int. 2008. PMID: 18343166
-
SPD-2/CEP192 and CDK Are Limiting for Microtubule-Organizing Center Function at the Centrosome.Curr Biol. 2015 Jul 20;25(14):1924-31. doi: 10.1016/j.cub.2015.06.001. Epub 2015 Jun 25. Curr Biol. 2015. PMID: 26119750
-
The newly found functions of MTOC in immunological response.J Leukoc Biol. 2014 Mar;95(3):417-30. doi: 10.1189/jlb.0813468. Epub 2013 Dec 2. J Leukoc Biol. 2014. PMID: 24295827 Review.
Cited by
-
Inhibitory Activities of Rare Ginsenoside Rg4 on Cecal Ligation and Puncture-Induced Sepsis.Int J Mol Sci. 2022 Sep 16;23(18):10836. doi: 10.3390/ijms231810836. Int J Mol Sci. 2022. PMID: 36142743 Free PMC article.
-
Ginsenoside Rg4 Enhances the Inductive Effects of Human Dermal Papilla Spheres on Hair Growth Via the AKT/GSK-3β/β-Catenin Signaling Pathway.J Microbiol Biotechnol. 2021 Jul 28;31(7):933-941. doi: 10.4014/jmb.2101.01032. J Microbiol Biotechnol. 2021. PMID: 34099599 Free PMC article.
-
Ginsenosides: changing the basic hallmarks of cancer cells to achieve the purpose of treating breast cancer.Chin Med. 2023 Sep 25;18(1):125. doi: 10.1186/s13020-023-00822-9. Chin Med. 2023. PMID: 37749560 Free PMC article. Review.
-
Natural products in attenuating renal inflammation via inhibiting the NLRP3 inflammasome in diabetic kidney disease.Front Immunol. 2023 May 5;14:1196016. doi: 10.3389/fimmu.2023.1196016. eCollection 2023. Front Immunol. 2023. PMID: 37215100 Free PMC article. Review.
-
Rg6, a rare ginsenoside, inhibits systemic inflammation through the induction of interleukin-10 and microRNA-146a.Sci Rep. 2019 Mar 13;9(1):4342. doi: 10.1038/s41598-019-40690-8. Sci Rep. 2019. PMID: 30867482 Free PMC article.
References
-
- Chida Y, Hamer M, Wardle J, Steptoe A. Do stress-related psychosocial factors contribute to cancer incidence and survival? Nat. Clin. Pract. Oncol. 2008;5:466–475. - PubMed
-
- Rudolph KL, Chang S, Lee HW, Blasco M, Gottlieb GJ, Greider C, DePinho RA. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell. 1999;96:701–712. - PubMed
-
- Tsigos C, Chrousos GP. Hypothalamic-pituitary-adrenal axis, neuroendocrine factors and stress. J. Psychosom. Res. 2002;53:865–871. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
