

Albumin contributes to kidney disease progression in Alport syndrome
- PMID: 27147675
- PMCID: PMC4967167
- DOI: 10.1152/ajprenal.00456.2015
Albumin contributes to kidney disease progression in Alport syndrome
Abstract
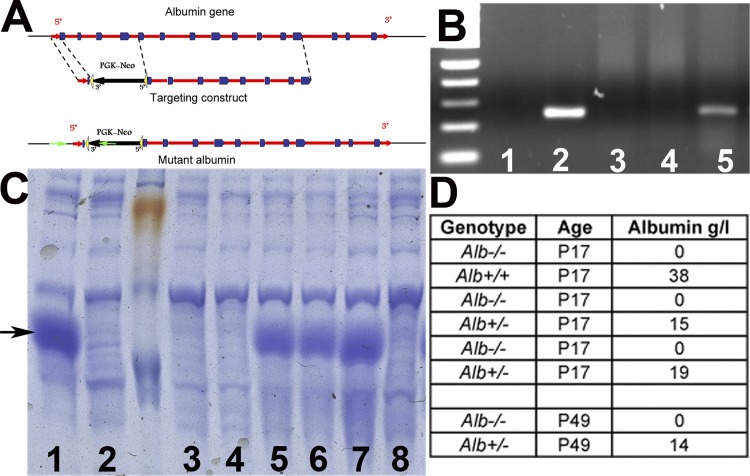
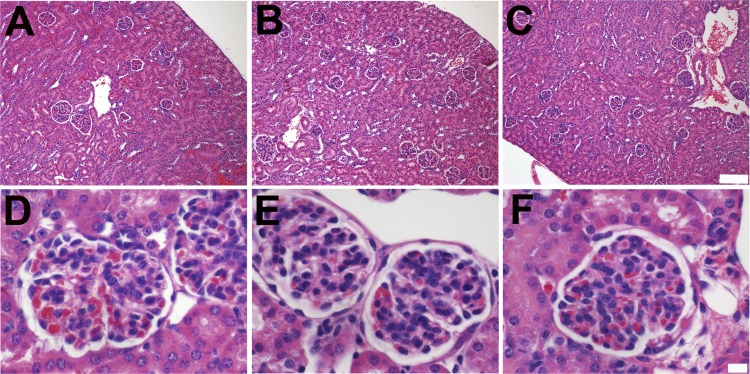
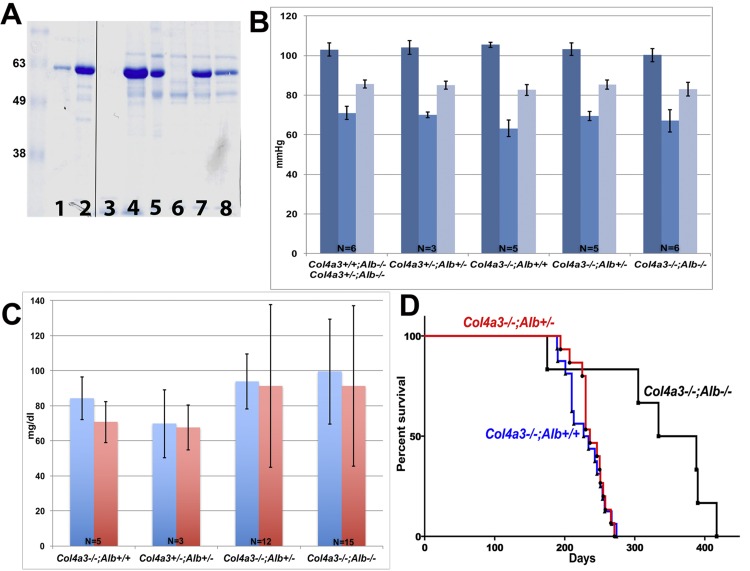
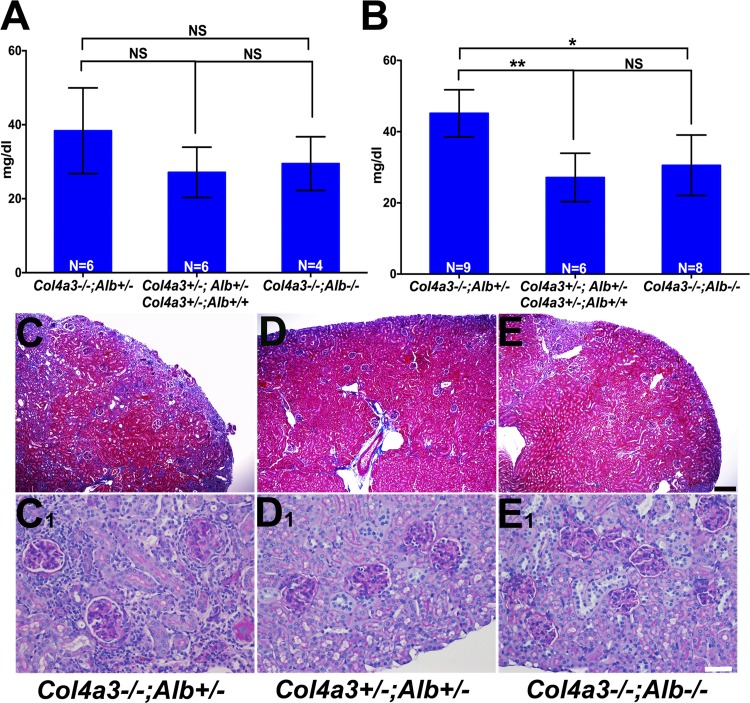
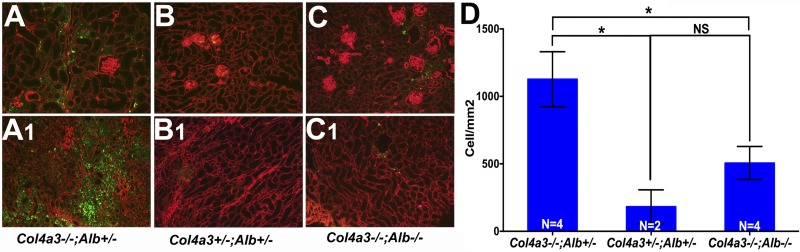
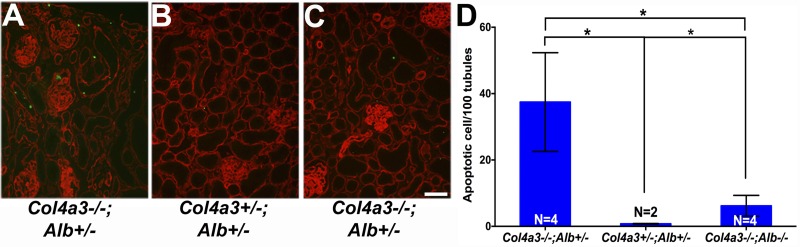
Alport syndrome is a familial kidney disease caused by defects in the collagen type IV network of the glomerular basement membrane. Lack of collagen-α3α4α5(IV) changes the glomerular basement membrane morphologically and functionally, rendering it leaky to albumin and other plasma proteins. Filtered albumin has been suggested to be a cause of the glomerular and tubular injuries observed at advanced stages of Alport syndrome. To directly investigate the role that albumin plays in the progression of disease in Alport syndrome, we generated albumin knockout (Alb(-/-)) mice to use as a tool for removing albuminuria as a component of kidney disease. Mice lacking albumin were healthy and indistinguishable from control littermates, although they developed hypertriglyceridemia. Dyslipidemia was observed in Alb(+/-) mice, which displayed half the normal plasma albumin concentration. Alb mutant mice were bred to collagen-α3(IV) knockout (Col4a3(-/-)) mice, which are a model for human Alport syndrome. Lack of circulating and filtered albumin in Col4a3(-/-);Alb(-/-) mice resulted in dramatically improved kidney disease outcomes, as these mice lived 64% longer than did Col4a3(-/-);Alb(+/+) and Col4a3(-/-);Alb(+/-) mice, despite similar blood pressures and serum triglyceride levels. Further investigations showed that the absence of albumin correlated with reduced transforming growth factor-β1 signaling as well as reduced tubulointerstitial, glomerular, and podocyte pathology. We conclude that filtered albumin is injurious to kidney cells in Alport syndrome and perhaps in other proteinuric kidney diseases, including diabetic nephropathy.
Keywords: Alport syndrome; albumin; collagen type IV.
Copyright © 2016 the American Physiological Society.
Figures
References
-
- Abbate M, Zoja C, Morigi M, Rottoli D, Angioletti S, Tomasoni S, Zanchi C, Longaretti L, Donadelli R, Remuzzi G. Transforming growth factor-β1 is up-regulated by podocytes in response to excess intraglomerular passage of proteins: a central pathway in progressive glomerulosclerosis. Am J Pathol 161: 2179–2193, 2002. - PMC - PubMed
-
- Abe H, Shibuya T, Odashima S, Arichi S, Nagase S. Alterations in the glomerulus in aminonucleoside nephrosis in analbuminemic rats. Nephron 50: 351–355, 1988. - PubMed
-
- Abrahamson DR, Isom K, Roach E, Stroganova L, Zelenchuk A, Miner JH, St John PL. Laminin compensation in collagen α3(IV) knockout (Alport) glomeruli contributes to permeability defects. J Am Soc Nephrol 18: 2465–2472, 2007. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous