

Characterization of the Gut Microbiome Using 16S or Shotgun Metagenomics
- PMID: 27148170
- PMCID: PMC4837688
- DOI: 10.3389/fmicb.2016.00459
Characterization of the Gut Microbiome Using 16S or Shotgun Metagenomics
Abstract
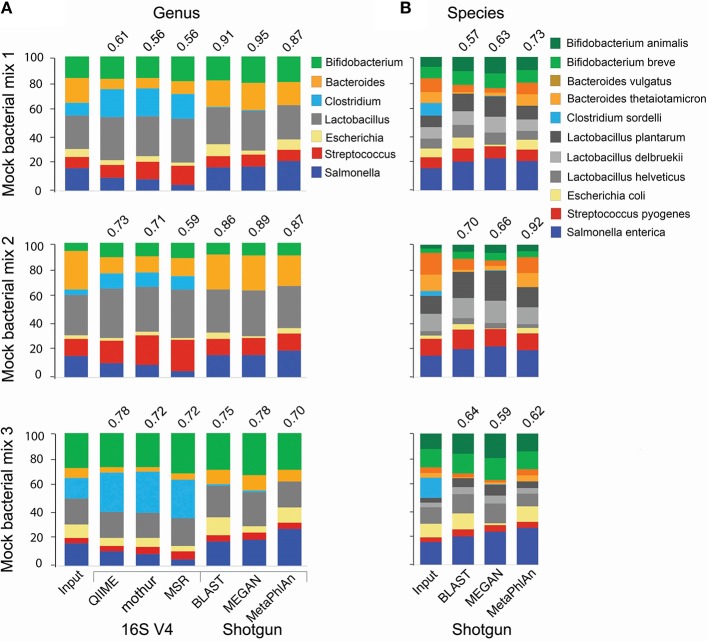
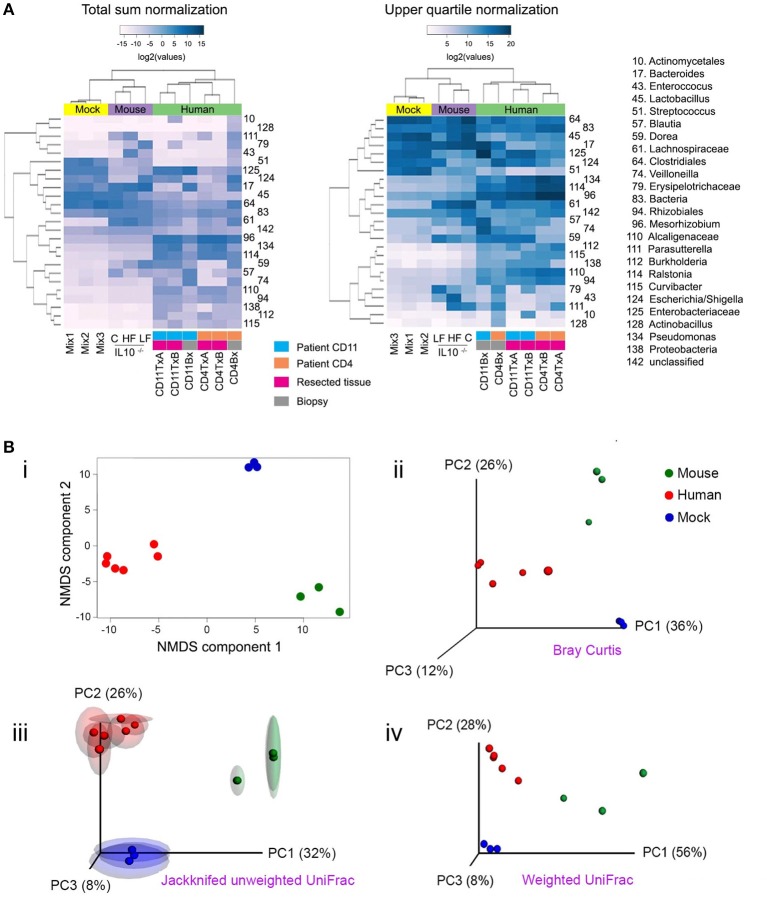
The advent of next generation sequencing (NGS) has enabled investigations of the gut microbiome with unprecedented resolution and throughput. This has stimulated the development of sophisticated bioinformatics tools to analyze the massive amounts of data generated. Researchers therefore need a clear understanding of the key concepts required for the design, execution and interpretation of NGS experiments on microbiomes. We conducted a literature review and used our own data to determine which approaches work best. The two main approaches for analyzing the microbiome, 16S ribosomal RNA (rRNA) gene amplicons and shotgun metagenomics, are illustrated with analyses of libraries designed to highlight their strengths and weaknesses. Several methods for taxonomic classification of bacterial sequences are discussed. We present simulations to assess the number of sequences that are required to perform reliable appraisals of bacterial community structure. To the extent that fluctuations in the diversity of gut bacterial populations correlate with health and disease, we emphasize various techniques for the analysis of bacterial communities within samples (α-diversity) and between samples (β-diversity). Finally, we demonstrate techniques to infer the metabolic capabilities of a bacteria community from these 16S and shotgun data.
Keywords: 16S rRNA gene sequencing; bioinformatics; diversity analysis; functional profiling; gut microbiome; shotgun metagenomics; taxonomic classification.
Figures



References
-
- Aho A., Hopcroft J., Ullman J. (1973). On finding lowest common ancestors in trees, in Proc. 5th ACM Symp. Theory of Computing (STOC), (New York, NY: ACM; ), 253–265.
-
- Aronesty E. (2011). Command-Line Tools for Processing Biological Sequencing Data ea-utils. Expression Analysis. Durham, NC: Available online at: http://code.google.com/p/ea-utils
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical