

The Origin and Activities of IgA1-Containing Immune Complexes in IgA Nephropathy
- PMID: 27148252
- PMCID: PMC4828451
- DOI: 10.3389/fimmu.2016.00117
The Origin and Activities of IgA1-Containing Immune Complexes in IgA Nephropathy
Abstract
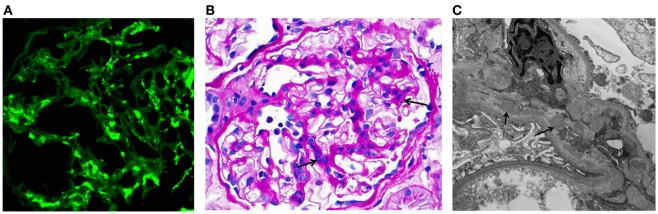
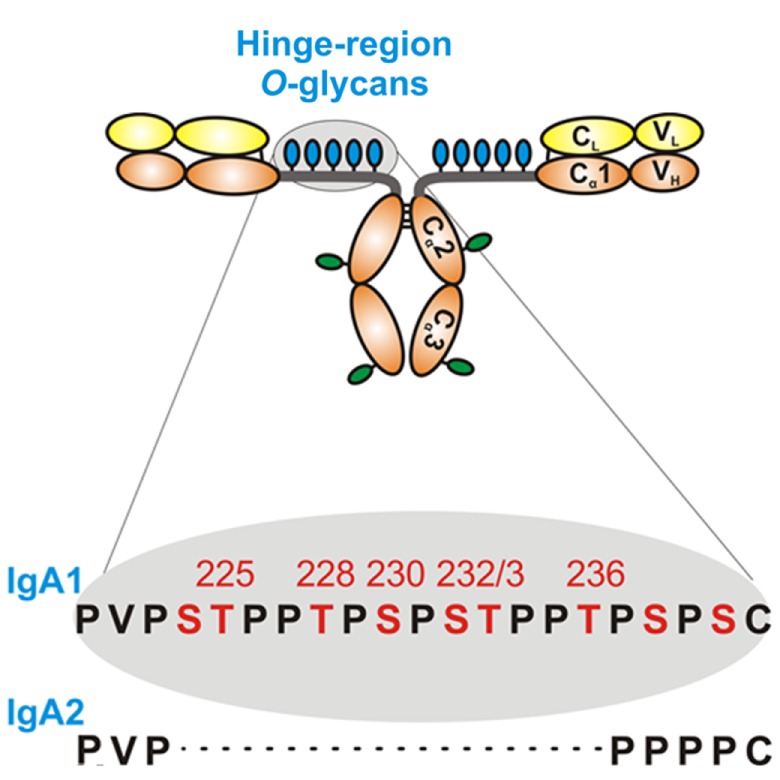
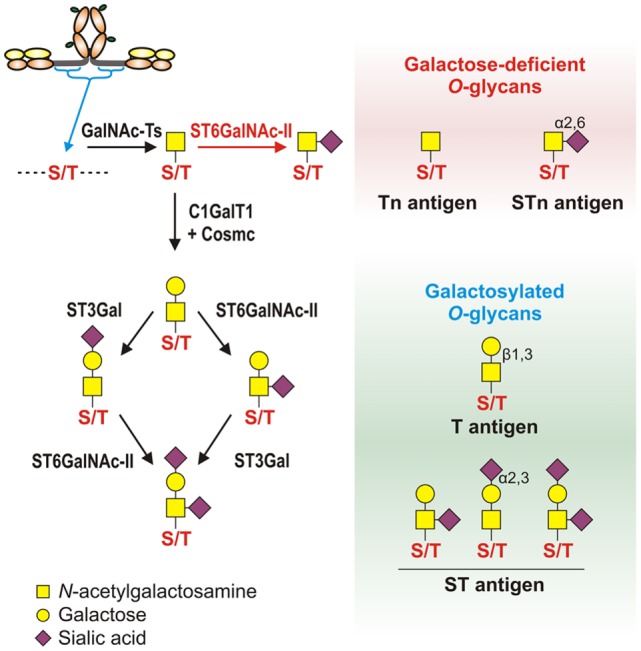
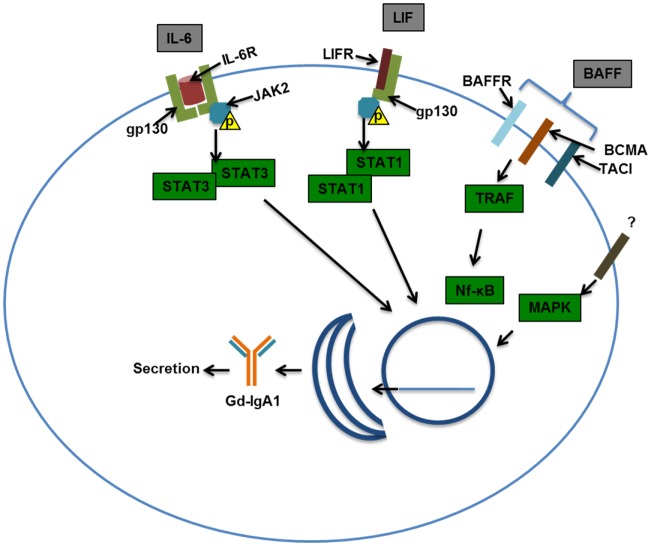
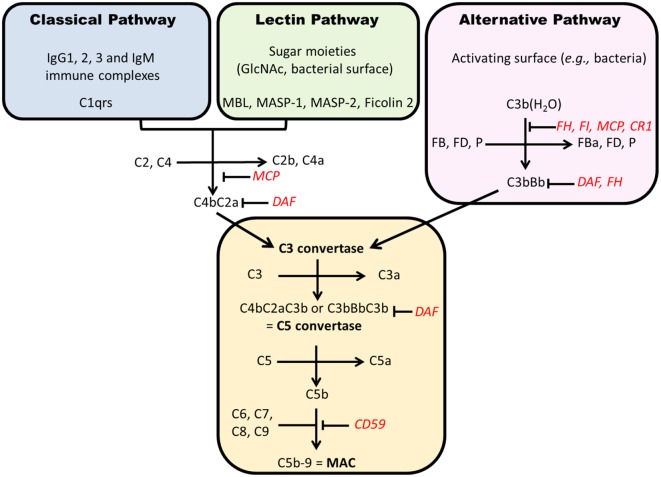
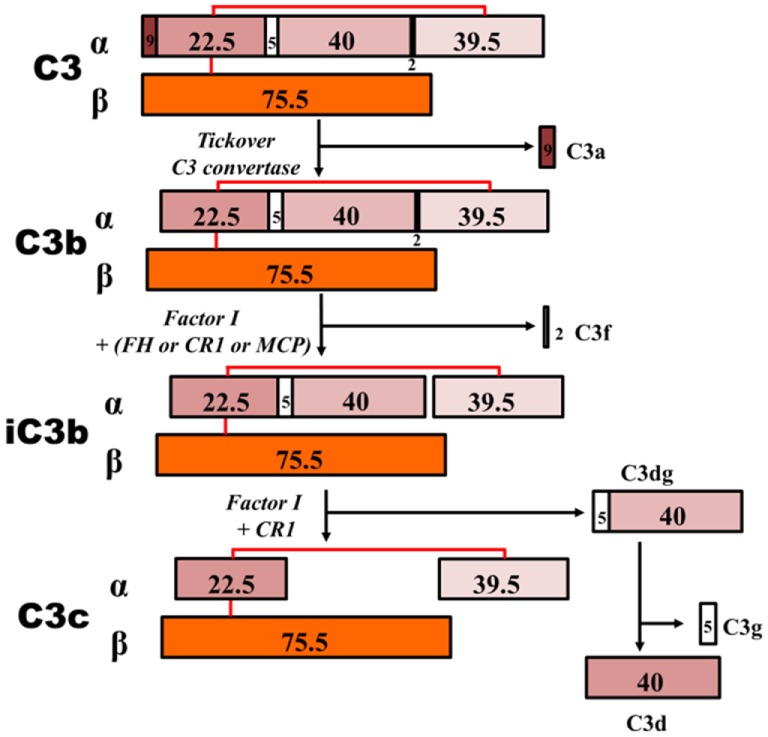
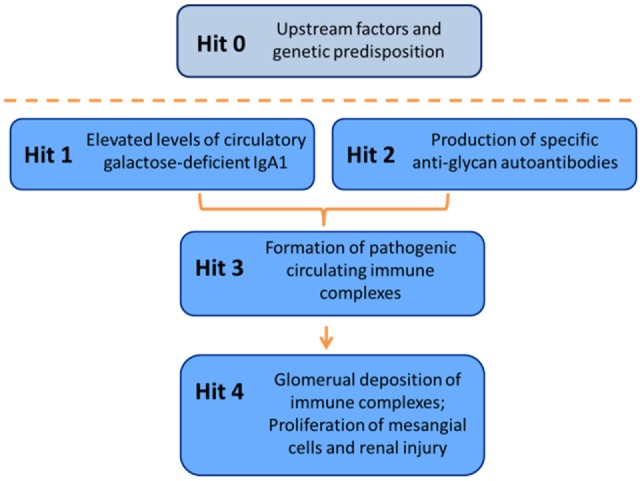
IgA nephropathy (IgAN) is the most common primary glomerulonephritis, frequently leading to end-stage renal disease, as there is no disease-specific therapy. IgAN is diagnosed from pathological assessment of a renal biopsy specimen based on predominant or codominant IgA-containing immunodeposits, usually with complement C3 co-deposits and with variable presence of IgG and/or IgM. The IgA in these renal deposits is galactose-deficient IgA1, with less than a full complement of galactose residues on the O-glycans in the hinge region of the heavy chains. Research from the past decade led to the definition of IgAN as an autoimmune disease with a multi-hit pathogenetic process with contributing genetic and environmental components. In this process, circulating galactose-deficient IgA1 (autoantigen) is bound by antiglycan IgG or IgA (autoantibodies) to form immune complexes. Some of these circulating complexes deposit in glomeruli, and thereby activate mesangial cells and induce renal injury through cellular proliferation and overproduction of extracellular matrix components and cytokines/chemokines. Glycosylation pathways associated with production of the autoantigen and the unique characteristics of the corresponding autoantibodies in patients with IgAN have been uncovered. Complement likely plays a significant role in the formation and the nephritogenic activities of these complexes. Complement activation is mediated through the alternative and lectin pathways and probably occurs systemically on IgA1-containing circulating immune complexes as well as locally in glomeruli. Incidence of IgAN varies greatly by geographical location; the disease is rare in central Africa but accounts for up to 40% of native-kidney biopsies in eastern Asia. Some of this variation may be explained by genetically determined influences on the pathogenesis of the disease. Genome-wide association studies to date have identified several loci associated with IgAN. Some of these loci are associated with the increased prevalence of IgAN, whereas others, such as deletion of complement factor H-related genes 1 and 3, are protective against the disease. Understanding the molecular mechanisms and genetic and biochemical factors involved in formation and activities of pathogenic IgA1-containing immune complexes will enable the development of future disease-specific therapies as well as identification of non-invasive disease-specific biomarkers.
Keywords: IgA; autoantibodies; complement C3; immune complexes; nephropathy.
Figures
References
-
- Berger J, Hinglais N. [Intercapillary deposits of IgA-IgG]. J Urol Nephrol (Paris) (1968) 74:694–5. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous