

A framework for understanding and targeting residual disease in oncogene-driven solid cancers
- PMID: 27149220
- PMCID: PMC5384713
- DOI: 10.1038/nm.4091
A framework for understanding and targeting residual disease in oncogene-driven solid cancers
Abstract
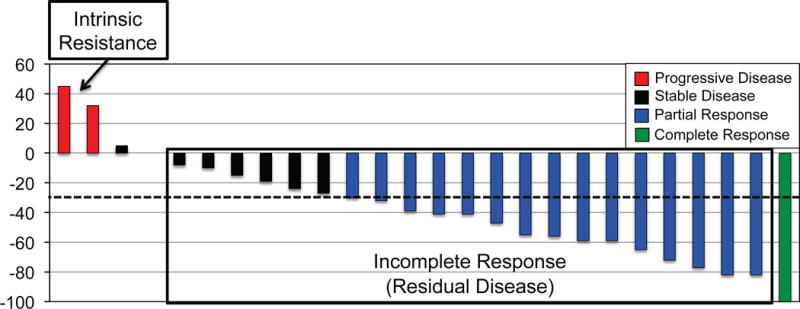
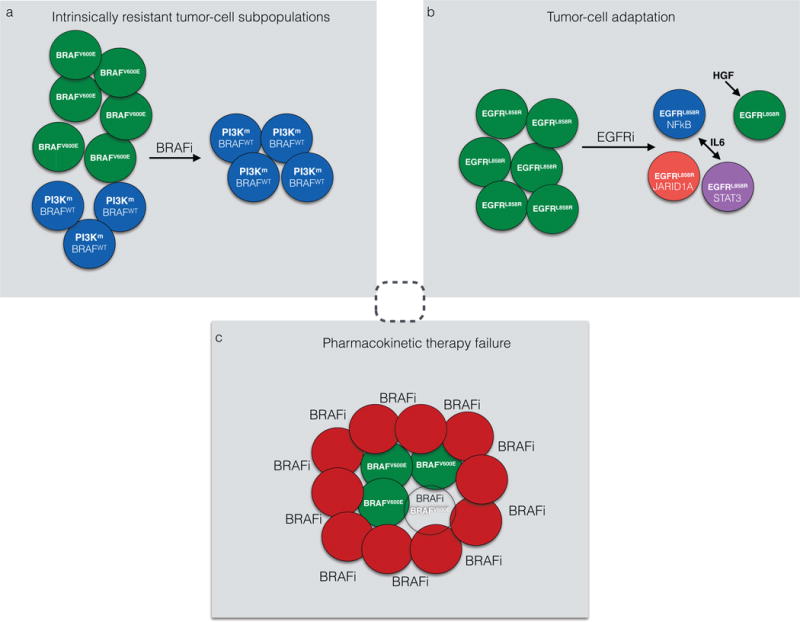
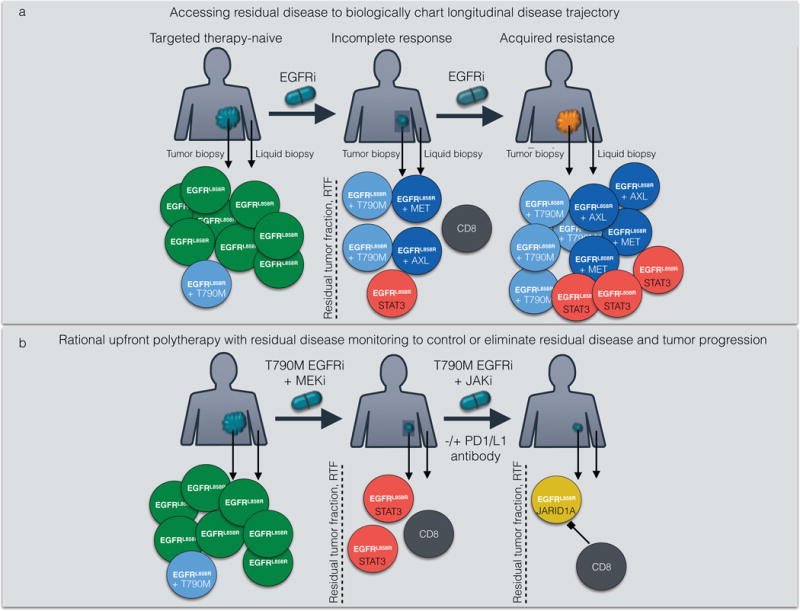
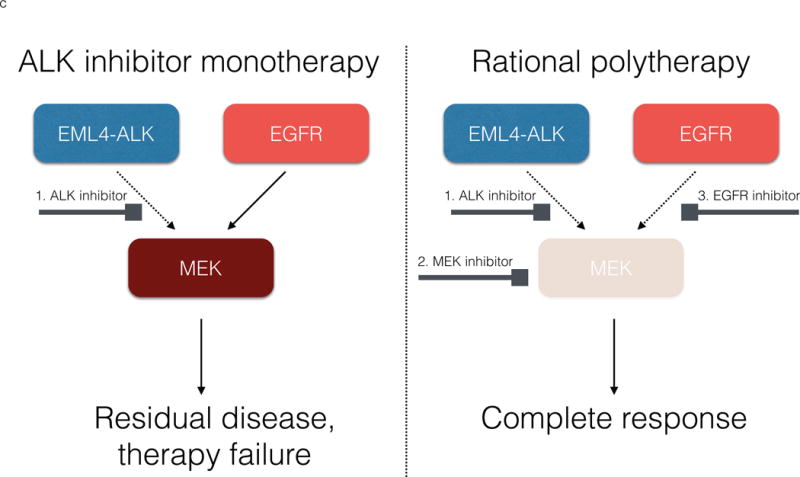
Molecular targeted therapy has the potential to dramatically improve survival in patients with cancer. However, complete and durable responses to targeted therapy are rare in individuals with advanced-stage solid cancers. Even the most effective targeted therapies generally do not induce a complete tumor response, resulting in residual disease and tumor progression that limits patient survival. We discuss the emerging need to more fully understand the molecular basis of residual disease as a prelude to designing therapeutic strategies to minimize or eliminate residual disease so that we can move from temporary to chronic control of disease, or a cure, for patients with advanced-stage solid cancers. Ultimately, we propose a shift from the current reactive paradigm of analyzing and treating acquired drug resistance to a pre-emptive paradigm of defining the mechanisms that result in residual disease, to target and limit this disease reservoir.
Conflict of interest statement
Figures
References
-
- Varmus H. Ten years on–the human genome and medicine. The New England journal of medicine. 2010;362:2028–2029. - PubMed
-
- Sawyers CL. Lessons learned from the development of kinase inhibitors. Clinical advances in hematology & oncology: H&O. 2009;7:588–589. - PubMed
-
- Sawyers CL. Shifting paradigms: the seeds of oncogene addiction. Nature medicine. 2009;15:1158–1161. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
