

Insulin Signaling in Insulin Resistance States and Cancer: A Modeling Analysis
- PMID: 27149630
- PMCID: PMC4858213
- DOI: 10.1371/journal.pone.0154415
Insulin Signaling in Insulin Resistance States and Cancer: A Modeling Analysis
Abstract
Insulin resistance is the common denominator of several diseases including type 2 diabetes and cancer, and investigating the mechanisms responsible for insulin signaling impairment is of primary importance. A mathematical model of the insulin signaling network (ISN) is proposed and used to investigate the dose-response curves of components of this network. Experimental data of C2C12 myoblasts with phosphatase and tensin homologue (PTEN) suppressed and data of L6 myotubes with induced insulin resistance have been analyzed by the model. We focused particularly on single and double Akt phosphorylation and pointed out insulin signaling changes related to insulin resistance. Moreover, a new characterization of the upstream signaling of the mammalian target of rapamycin complex 2 (mTORC2) is presented. As it is widely recognized that ISN proteins have a crucial role also in cell proliferation and death, the ISN model was linked to a cell population model and applied to data of a cell line of acute myeloid leukemia treated with a mammalian target of rapamycin inhibitor with antitumor activity. The analysis revealed simple relationships among the concentrations of ISN proteins and the parameters of the cell population model that characterize cell cycle progression and cell death.
Conflict of interest statement
Figures
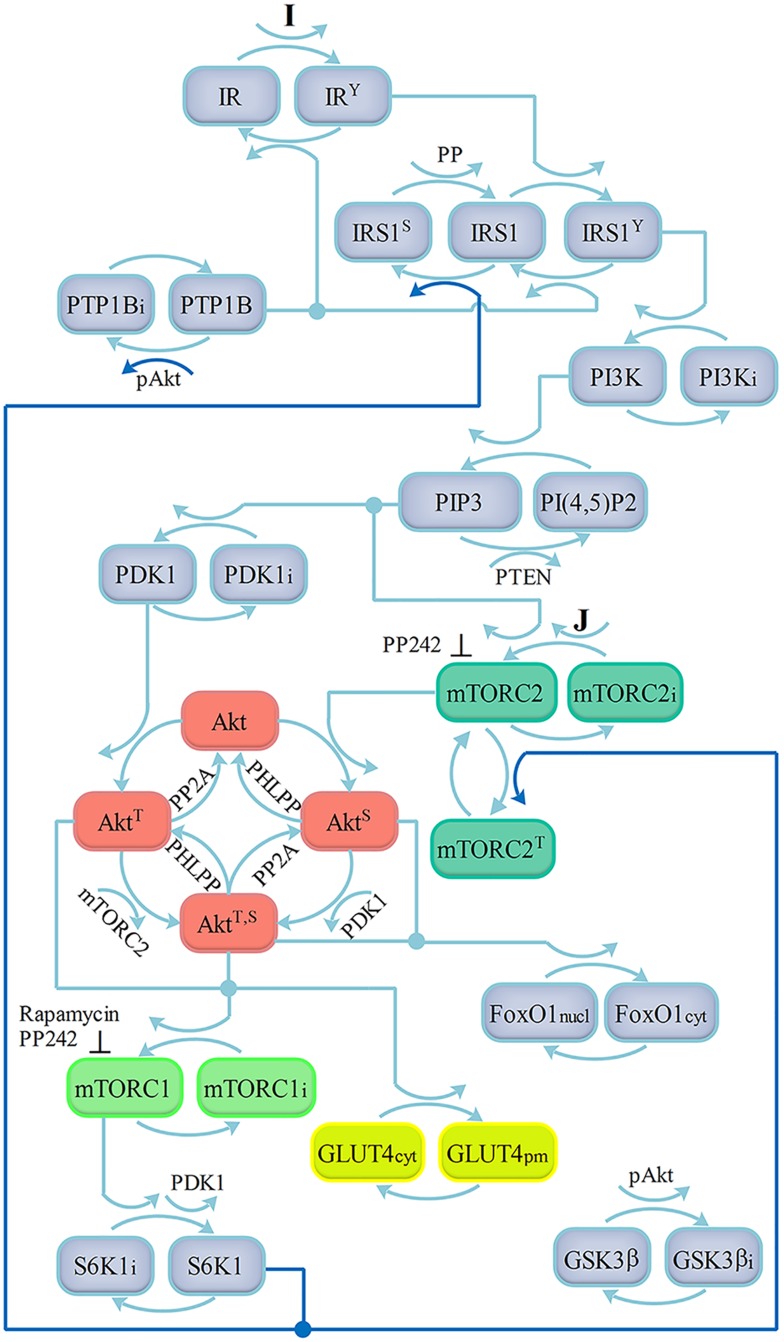
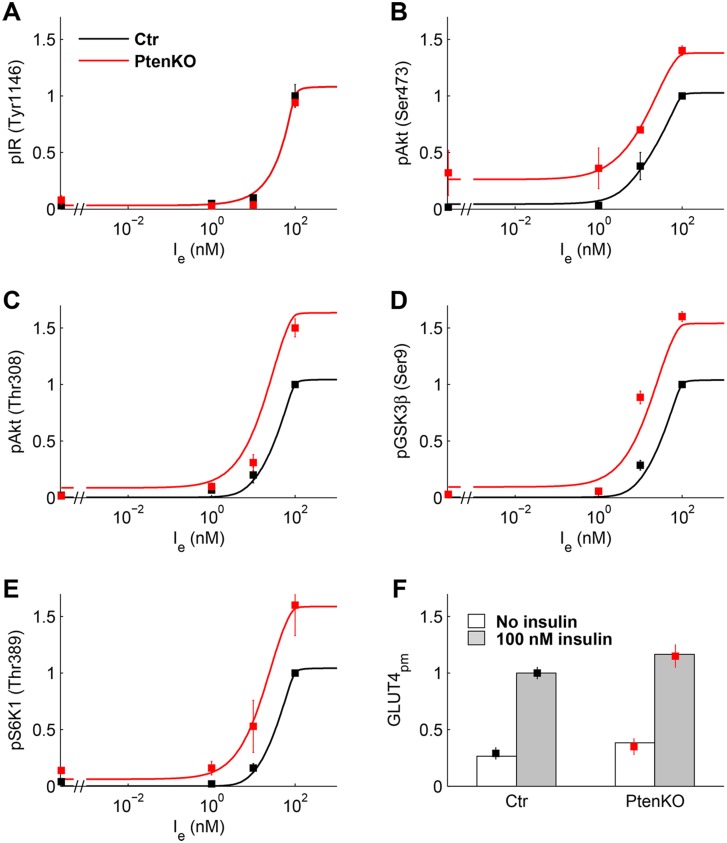
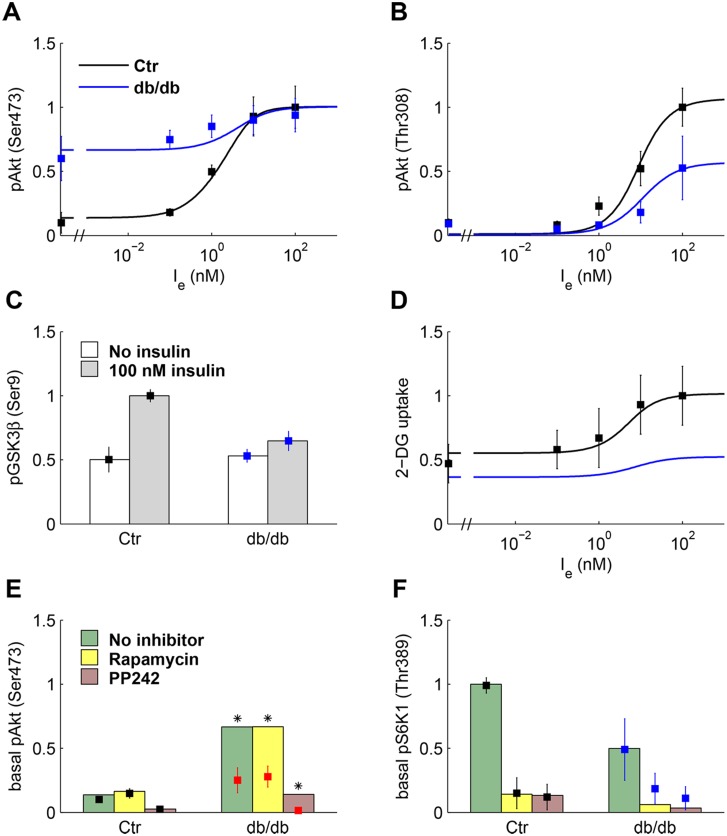
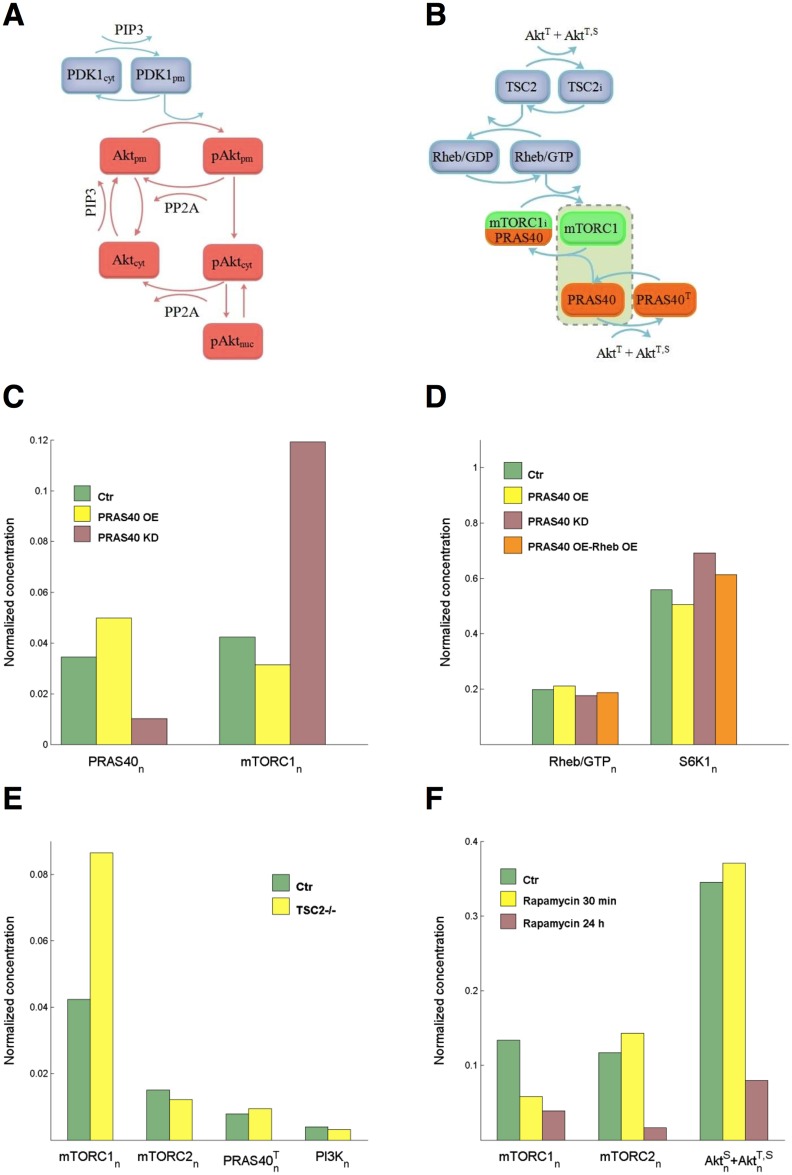
References
-
- Thong FS, Dugani CB, Klip A. Turning signals on and off: GLUT4 traffic in the insulin-signaling highway. Physiology (Bethesda). 2005; 20: 271–284. - PubMed
-
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005; 307(5712): 1098–1101. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
