

MNase titration reveals differences between nucleosome occupancy and chromatin accessibility
- PMID: 27151365
- PMCID: PMC4859066
- DOI: 10.1038/ncomms11485
MNase titration reveals differences between nucleosome occupancy and chromatin accessibility
Abstract
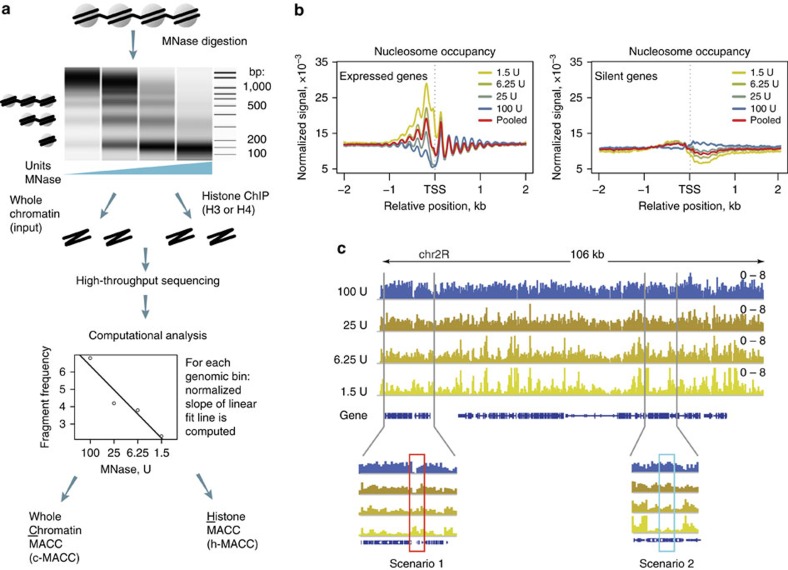
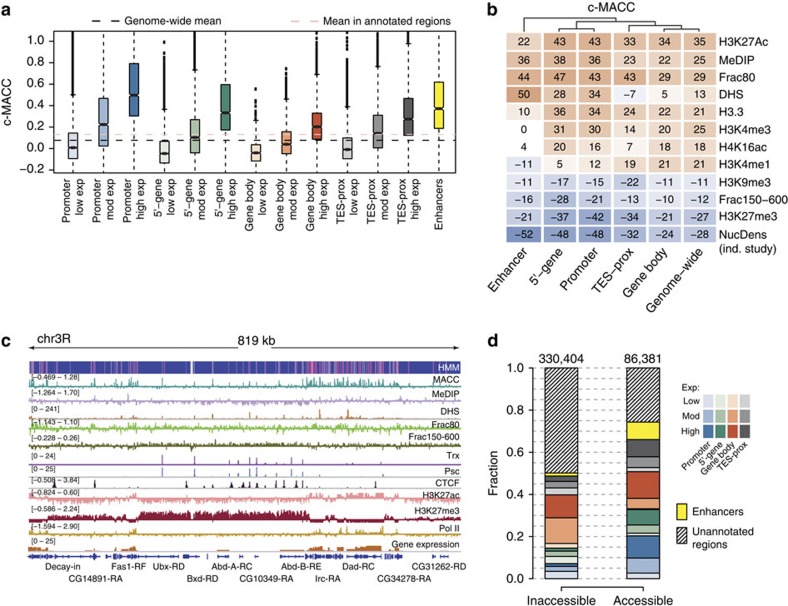
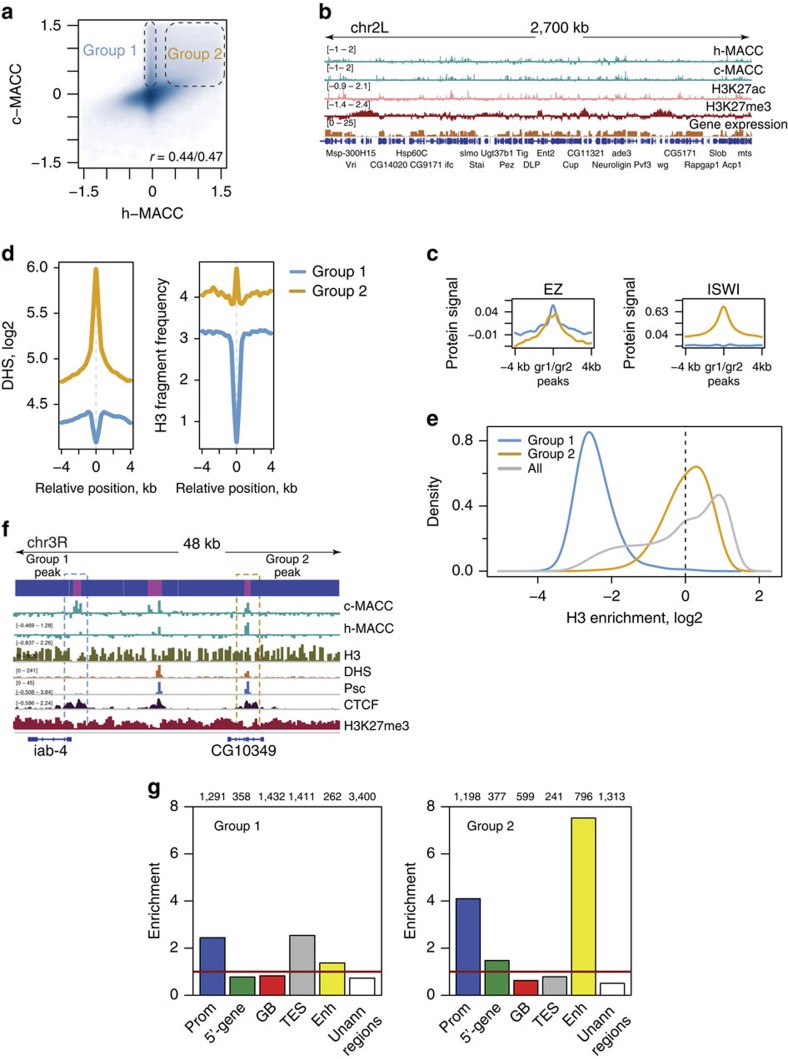
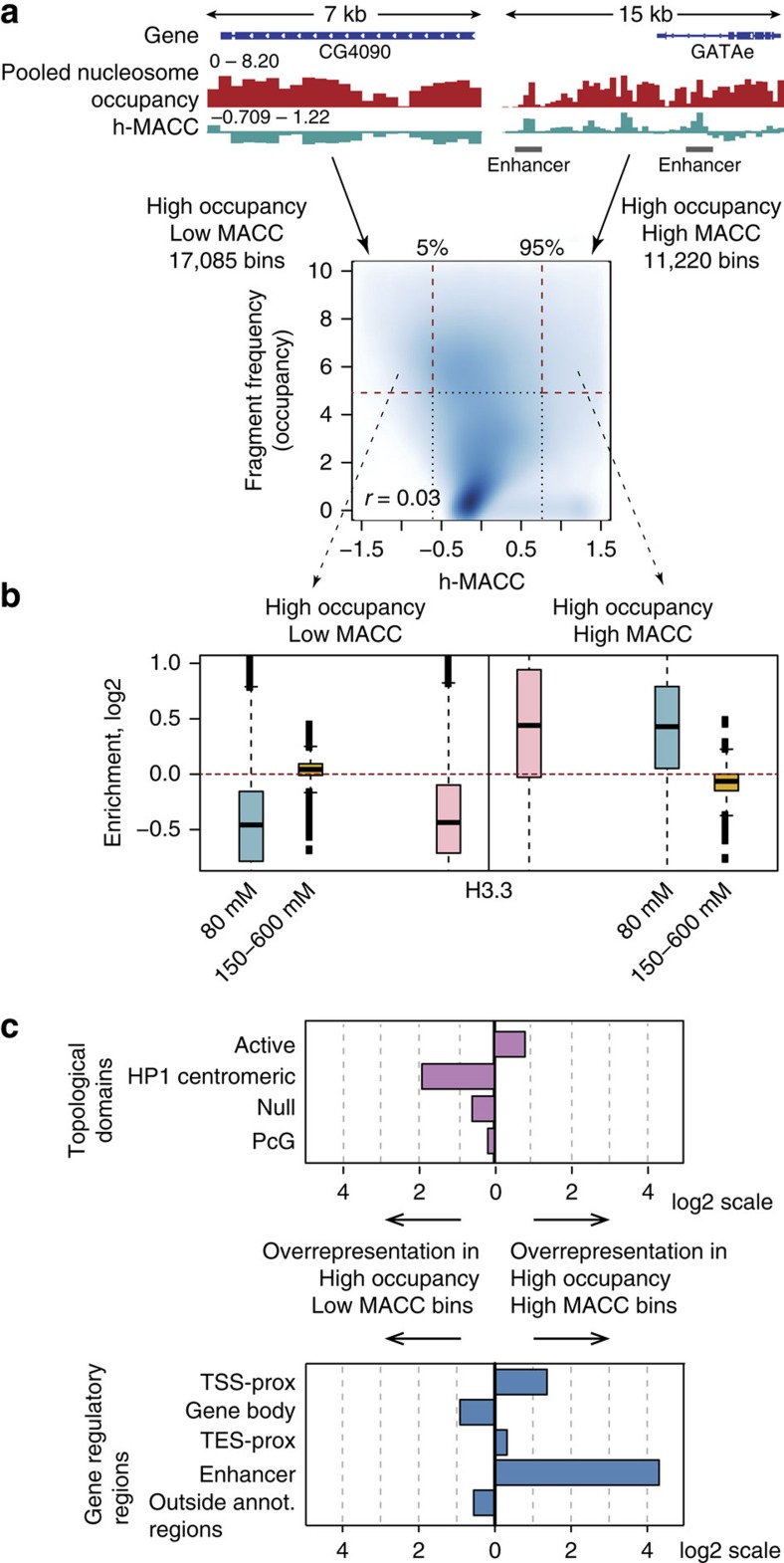
Chromatin accessibility plays a fundamental role in gene regulation. Nucleosome placement, usually measured by quantifying protection of DNA from enzymatic digestion, can regulate accessibility. We introduce a metric that uses micrococcal nuclease (MNase) digestion in a novel manner to measure chromatin accessibility by combining information from several digests of increasing depths. This metric, MACC (MNase accessibility), quantifies the inherent heterogeneity of nucleosome accessibility in which some nucleosomes are seen preferentially at high MNase and some at low MNase. MACC interrogates each genomic locus, measuring both nucleosome location and accessibility in the same assay. MACC can be performed either with or without a histone immunoprecipitation step, and thereby compares histone and non-histone protection. We find that changes in accessibility at enhancers, promoters and other regulatory regions do not correlate with changes in nucleosome occupancy. Moreover, high nucleosome occupancy does not necessarily preclude high accessibility, which reveals novel principles of chromatin regulation.
Figures
References
-
- Kornberg R. D. Chromatin structure: a repeating unit of histones and DNA. Science 184, 868–871 (1974). - PubMed
-
- Luger K., Mader A. W., Richmond R. K., Sargent D. F. & Richmond T. J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 251–260 (1997). - PubMed
-
- Wolffe A. P. & Brown D. D. Developmental regulation of two 5S ribosomal RNA genes. Science 241, 1626–1632 (1988). - PubMed
-
- Li B., Carey M. & Workman J. L. The role of chromatin during transcription. Cell 128, 707–719 (2007). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
