

Design of virus-based nanomaterials for medicine, biotechnology, and energy
- PMID: 27152673
- PMCID: PMC5068136
- DOI: 10.1039/c5cs00287g
Design of virus-based nanomaterials for medicine, biotechnology, and energy
Abstract
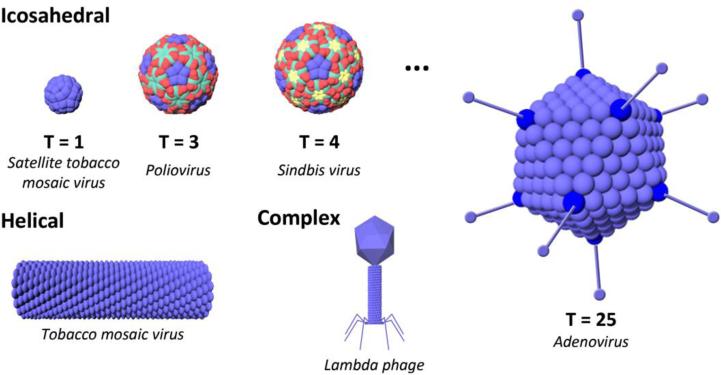
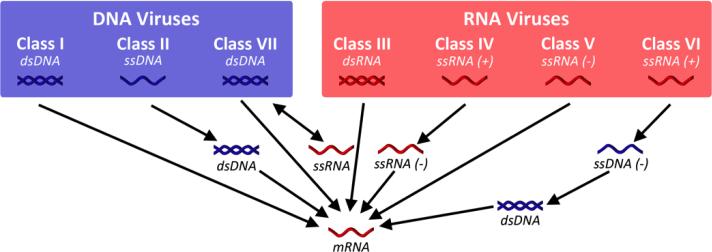
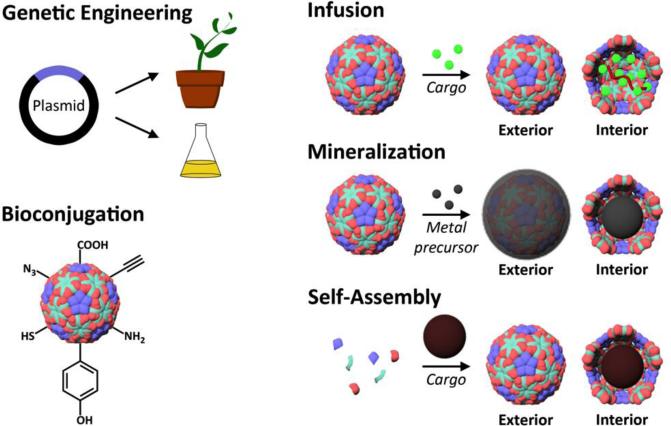
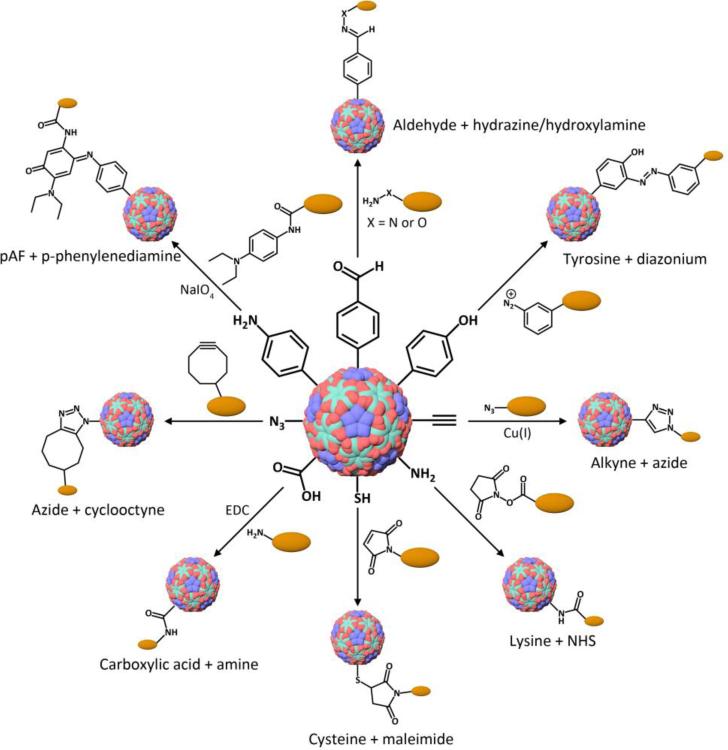
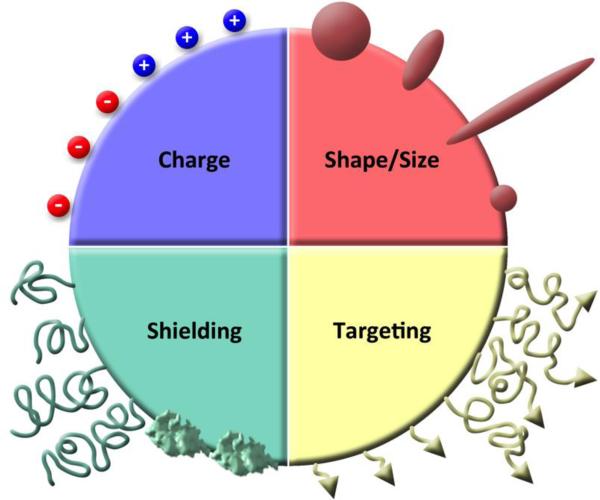
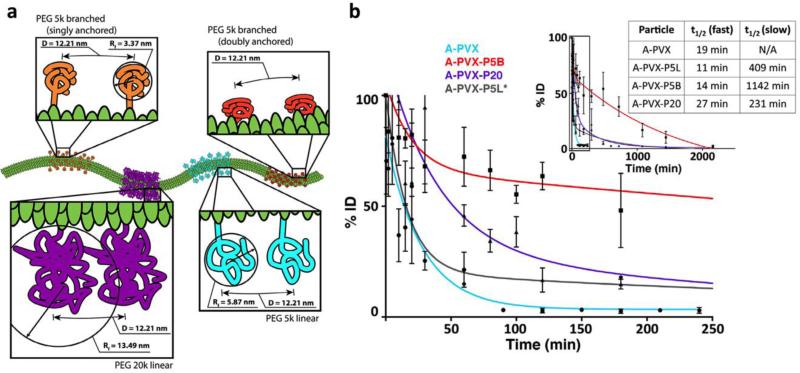
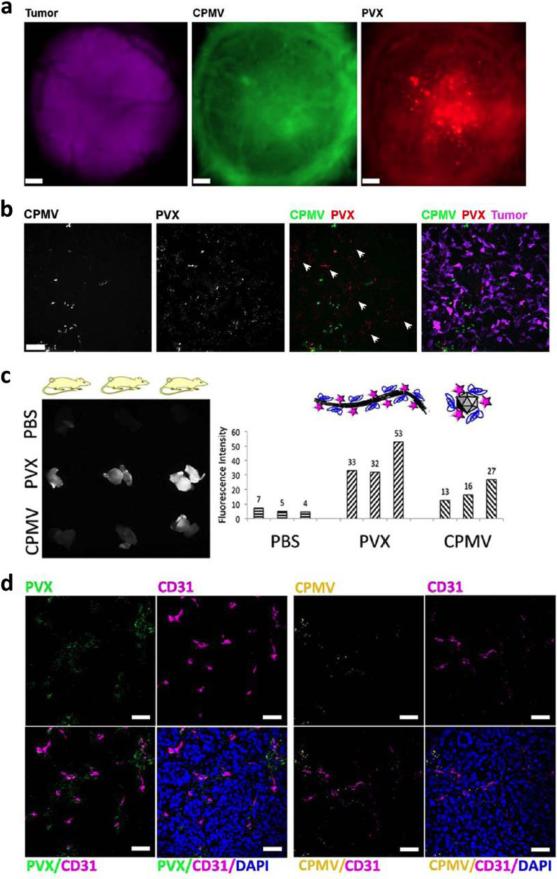
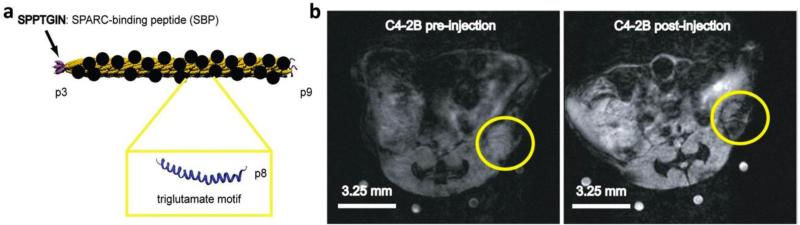
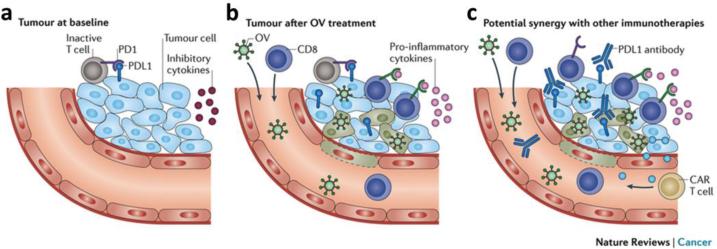
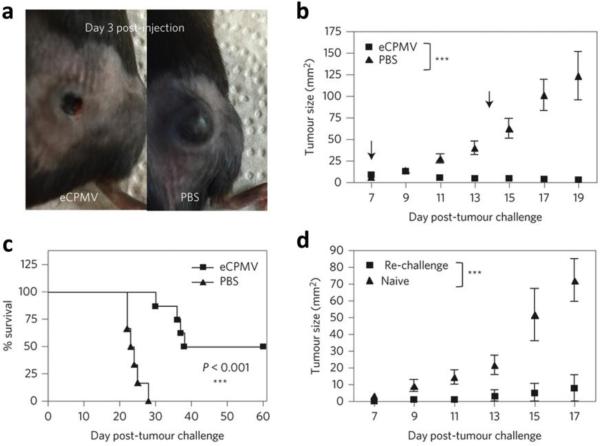
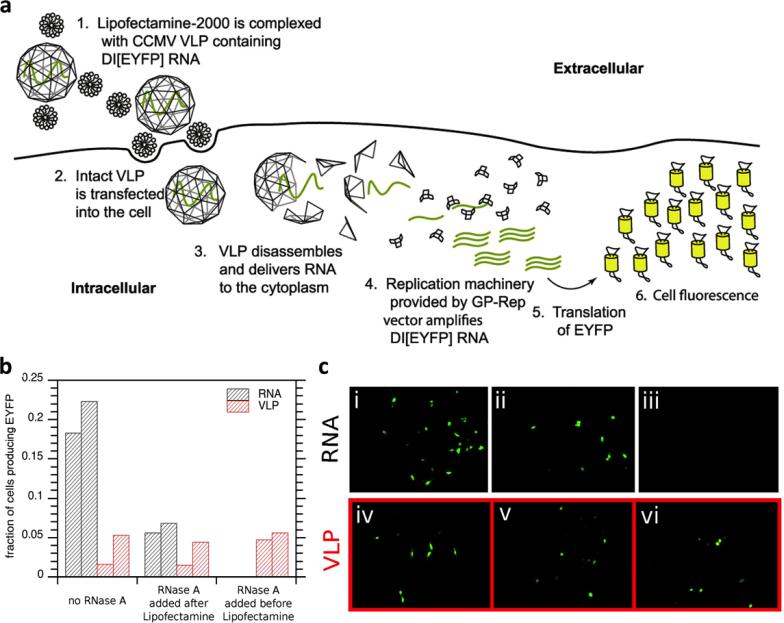
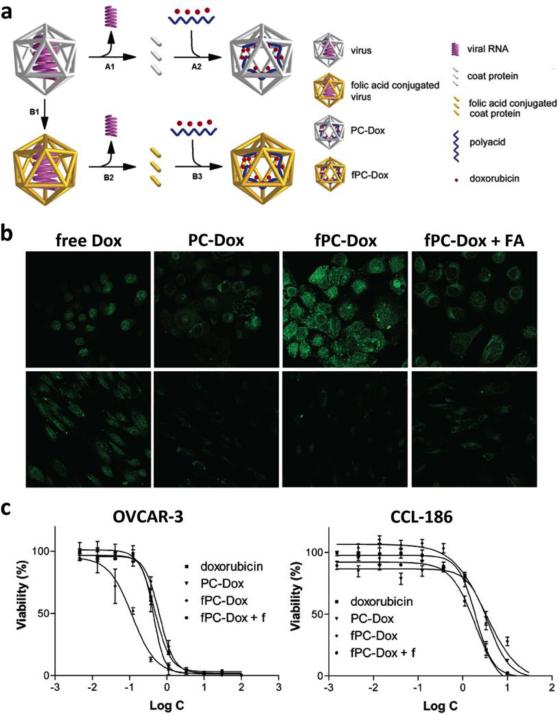
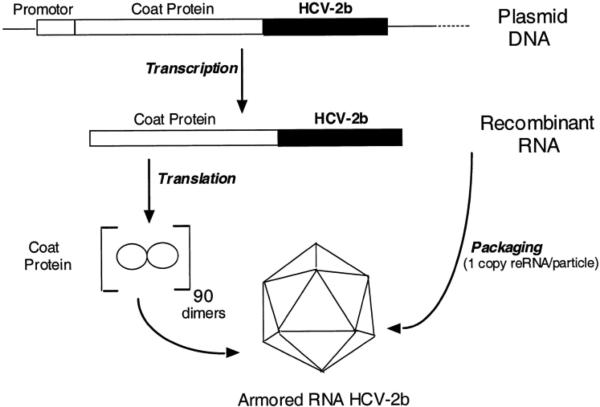
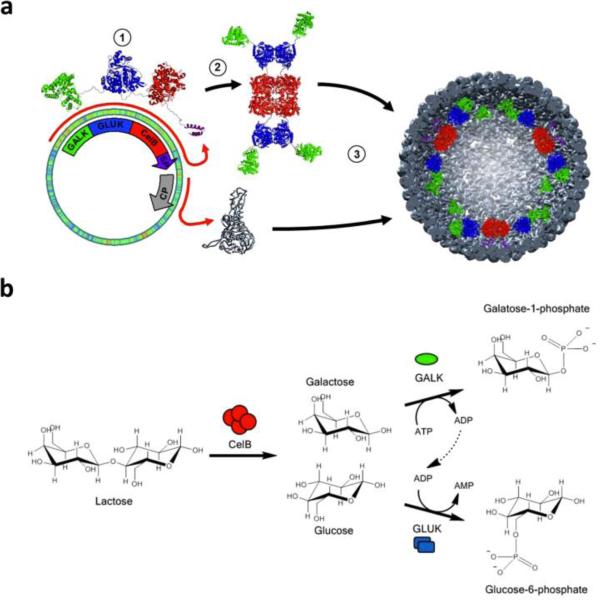
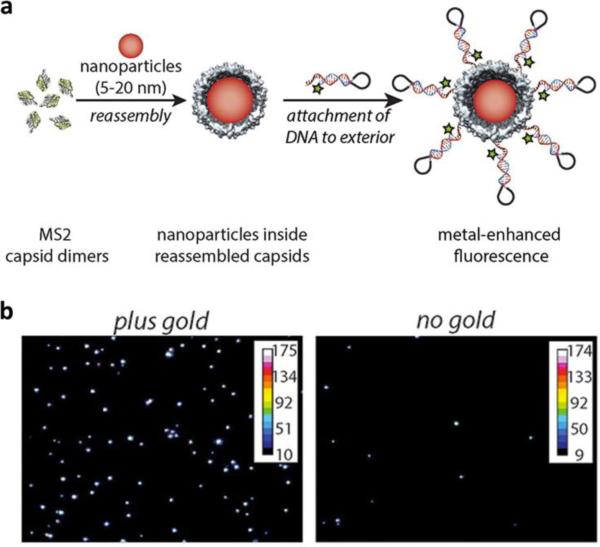
This review provides an overview of recent developments in "chemical virology." Viruses, as materials, provide unique nanoscale scaffolds that have relevance in chemical biology and nanotechnology, with diverse areas of applications. Some fundamental advantages of viruses, compared to synthetically programmed materials, include the highly precise spatial arrangement of their subunits into a diverse array of shapes and sizes and many available avenues for easy and reproducible modification. Here, we will first survey the broad distribution of viruses and various methods for producing virus-based nanoparticles, as well as engineering principles used to impart new functionalities. We will then examine the broad range of applications and implications of virus-based materials, focusing on the medical, biotechnology, and energy sectors. We anticipate that this field will continue to evolve and grow, with exciting new possibilities stemming from advancements in the rational design of virus-based nanomaterials.
Figures
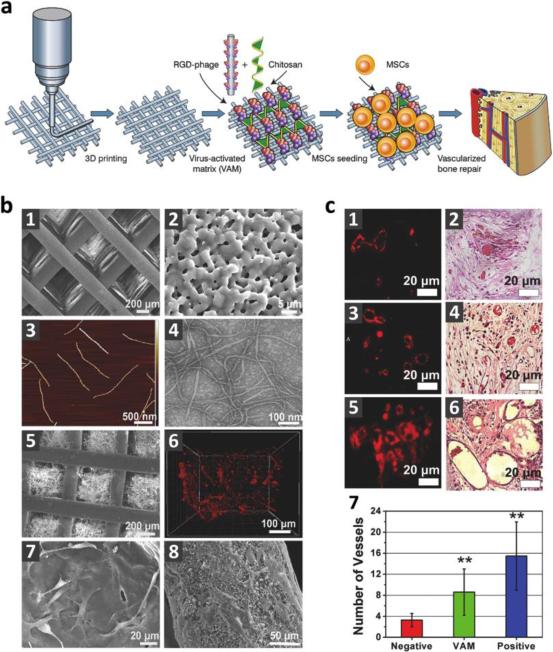
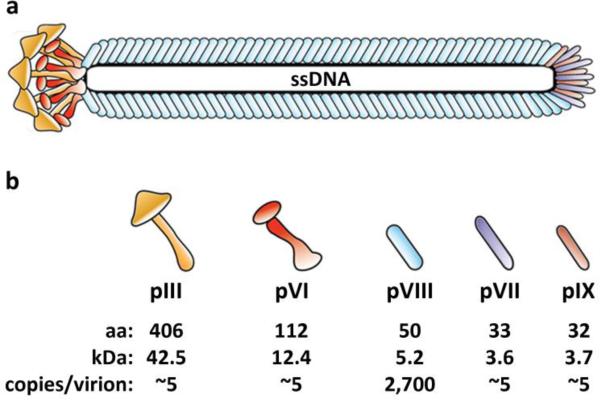
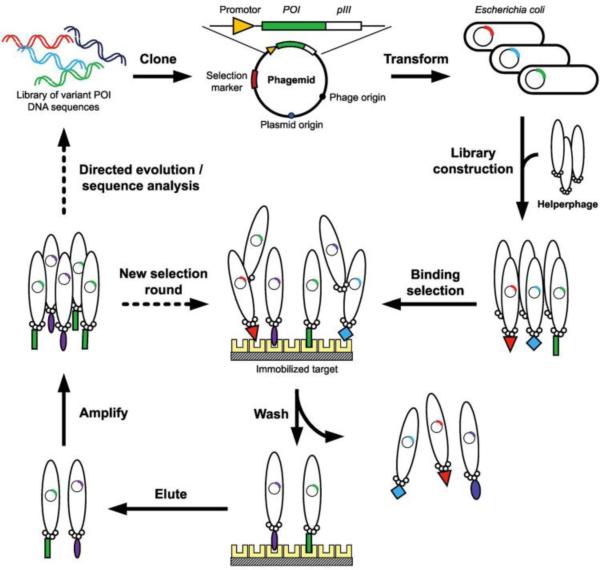
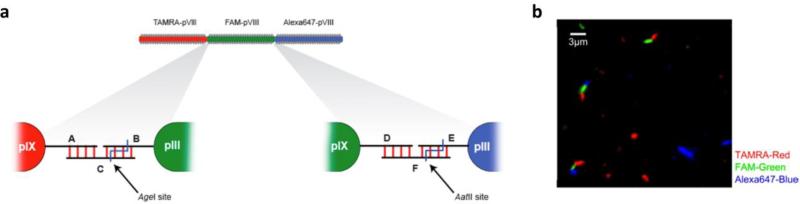
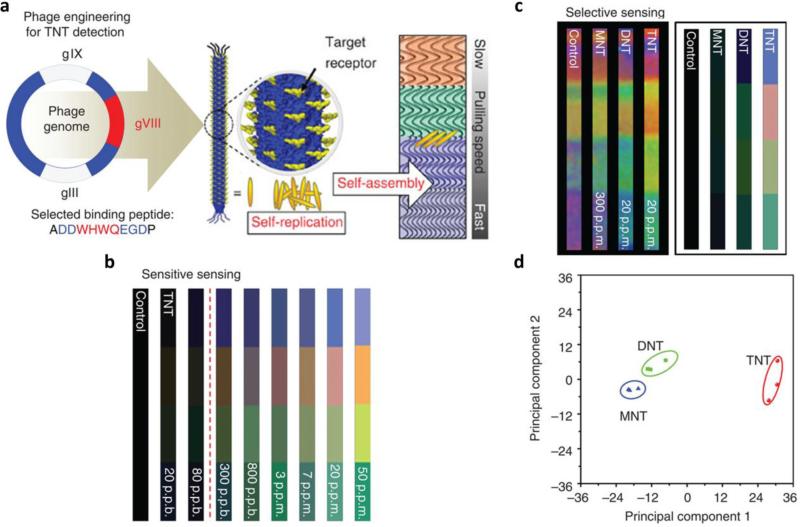

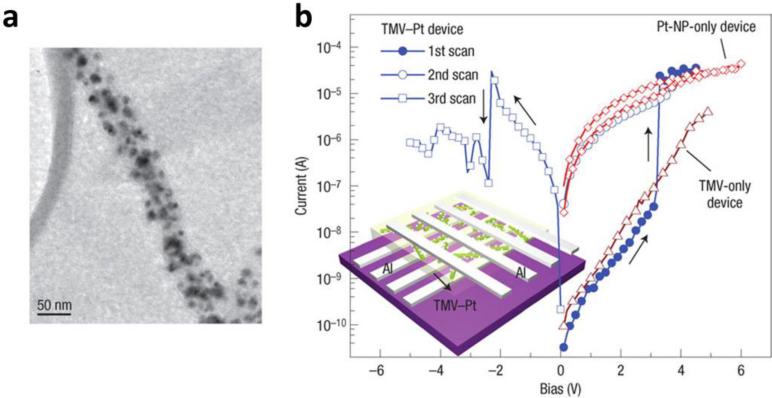
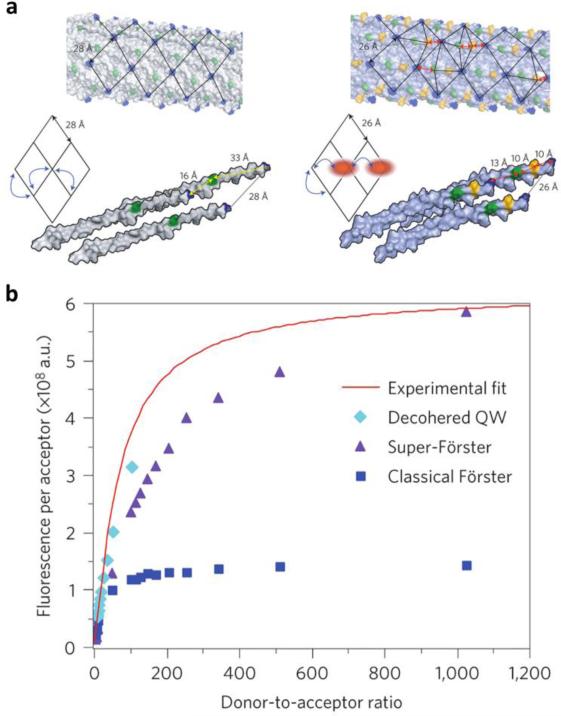
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources