

Temporal Stability of the Human Skin Microbiome
- PMID: 27153496
- PMCID: PMC4860256
- DOI: 10.1016/j.cell.2016.04.008
Temporal Stability of the Human Skin Microbiome
Abstract
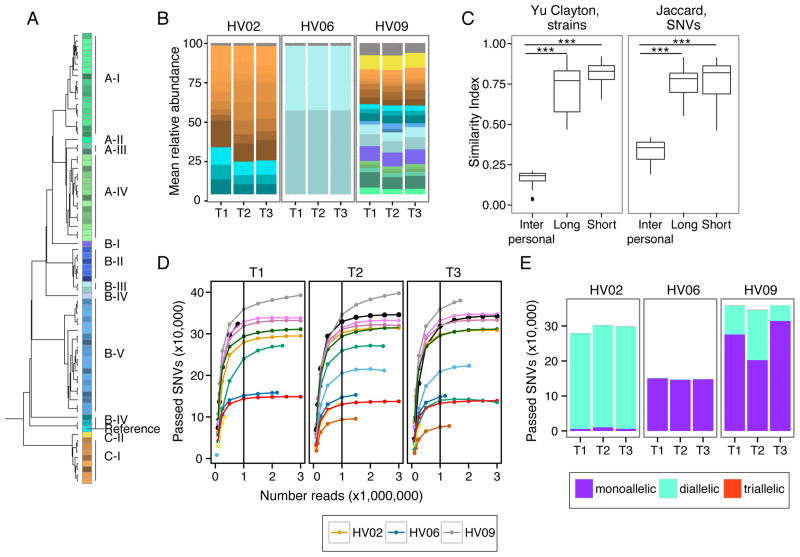
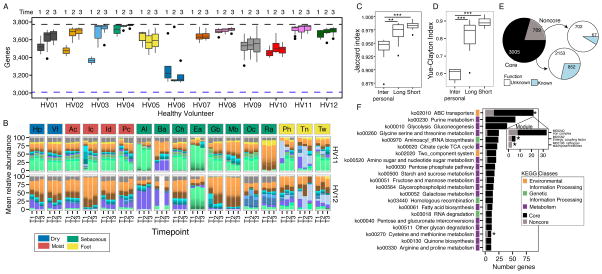
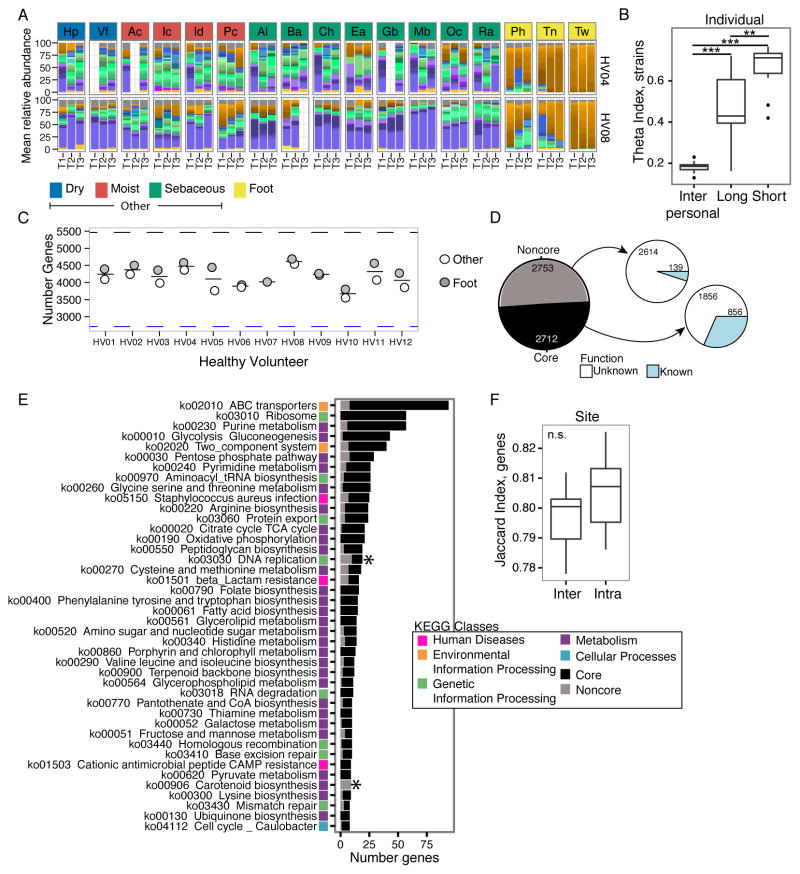
Biogeography and individuality shape the structural and functional composition of the human skin microbiome. To explore these factors' contribution to skin microbial community stability, we generated metagenomic sequence data from longitudinal samples collected over months and years. Analyzing these samples using a multi-kingdom, reference-based approach, we found that despite the skin's exposure to the external environment, its bacterial, fungal, and viral communities were largely stable over time. Site, individuality, and phylogeny were all determinants of stability. Foot sites exhibited the most variability; individuals differed in stability; and transience was a particular characteristic of eukaryotic viruses, which showed little site-specificity in colonization. Strain and single-nucleotide variant-level analysis showed that individuals maintain, rather than reacquire, prevalent microbes from the environment. Longitudinal stability of skin microbial communities generates hypotheses about colonization resistance and empowers clinical studies exploring alterations observed in disease states.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures
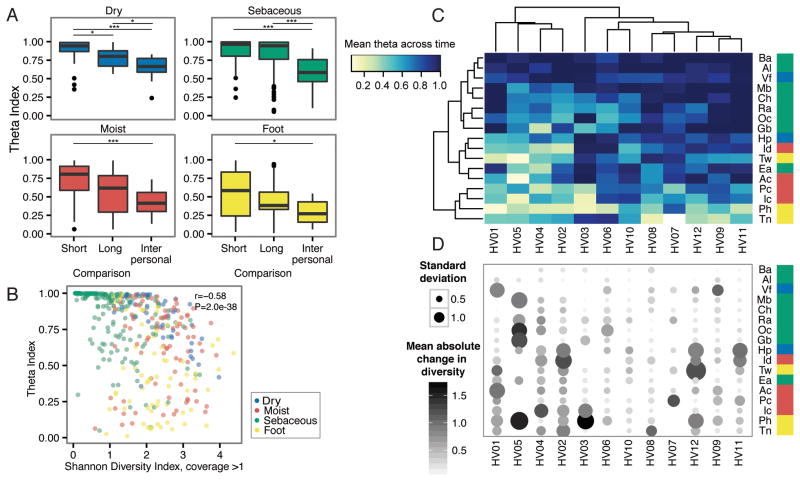
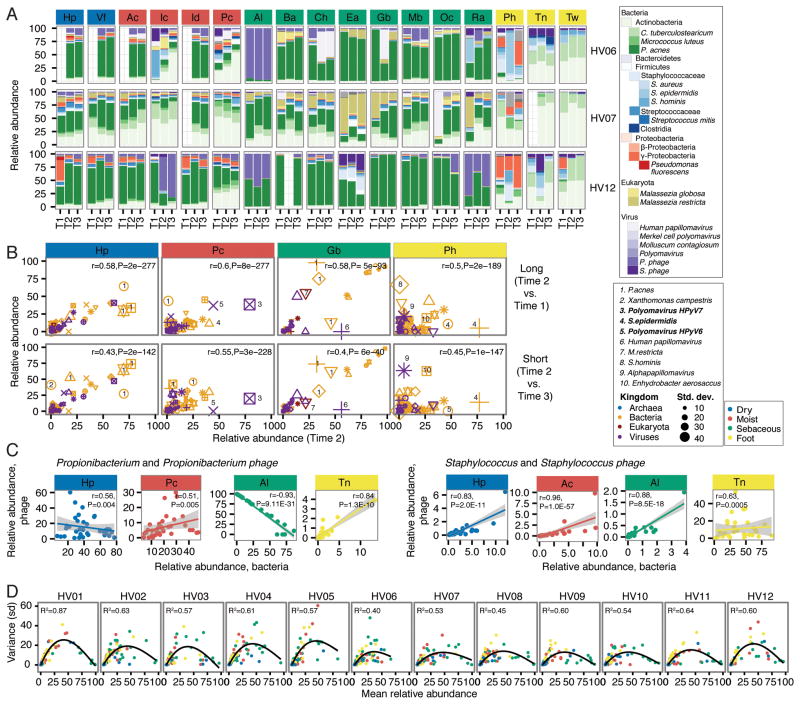
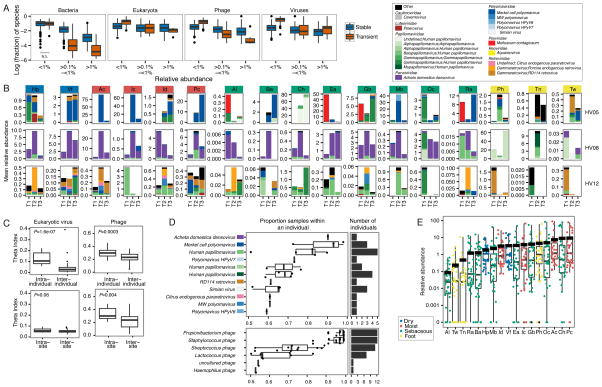
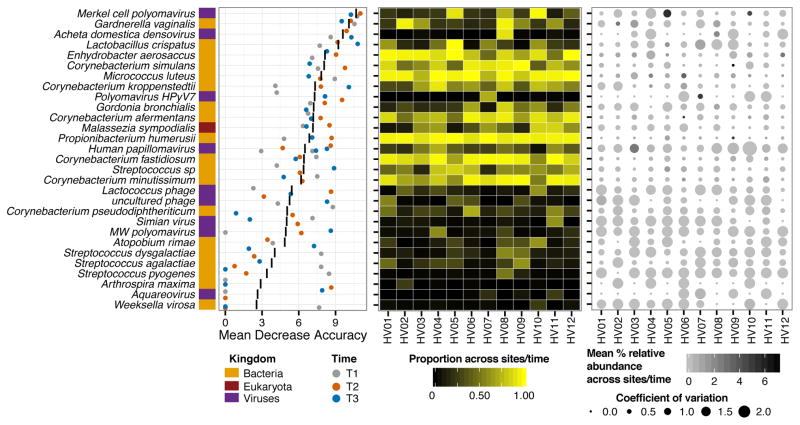
Comment in
-
Microbial Skin Inhabitants: Friends Forever.Cell. 2016 May 5;165(4):771-2. doi: 10.1016/j.cell.2016.04.035. Cell. 2016. PMID: 27153488
References
-
- Belkaid Y, Segre JA. Dialogue between skin microbiota and immunity. Science. 2014;346:954–959. - PubMed
-
- Conlan S, Mijares LA, Becker J, Blakesley RW, Bouffard GG, Brooks S, Coleman H, Gupta J, Gurson N, et al. NISC Comp Seq Program. Staphylococcus epidermidis pan-genome sequence analysis reveals diversity of skin commensal and hospital infection-associated isolates. Genome biology. 2012;13:R64. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
