

Remarkable evolutionary relatedness among the enzymes and proteins from the α-amylase family
- PMID: 27154042
- PMCID: PMC11108405
- DOI: 10.1007/s00018-016-2246-6
Remarkable evolutionary relatedness among the enzymes and proteins from the α-amylase family
Abstract
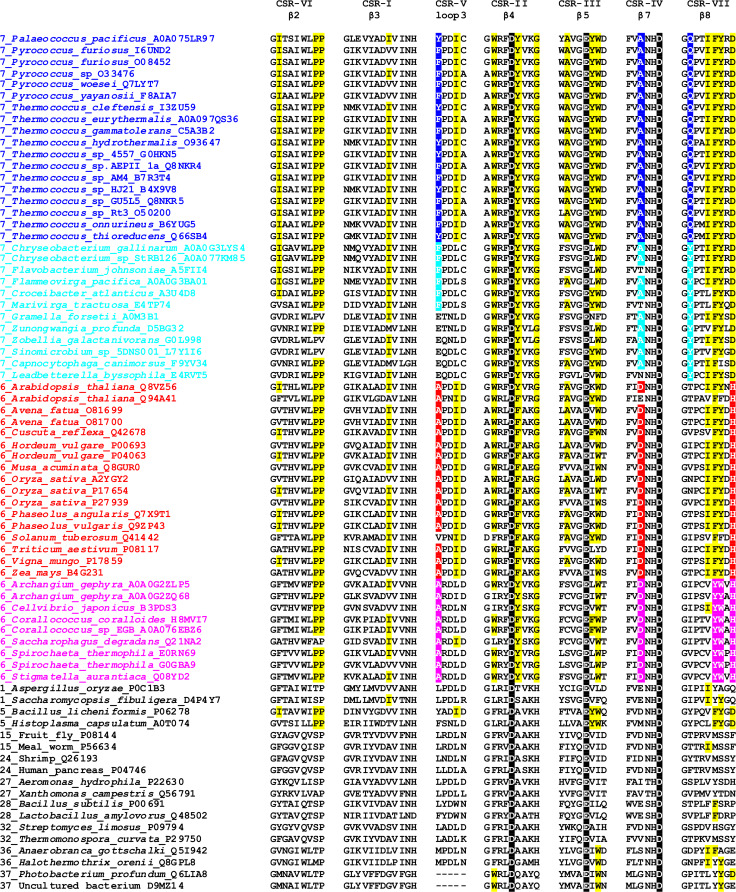
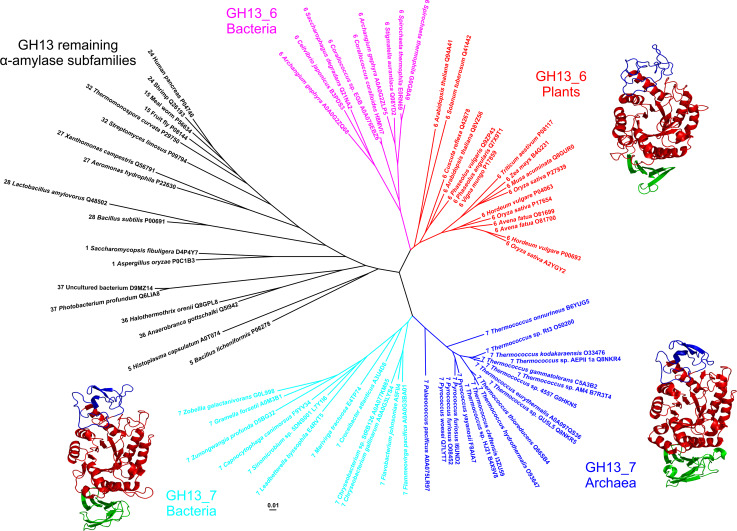
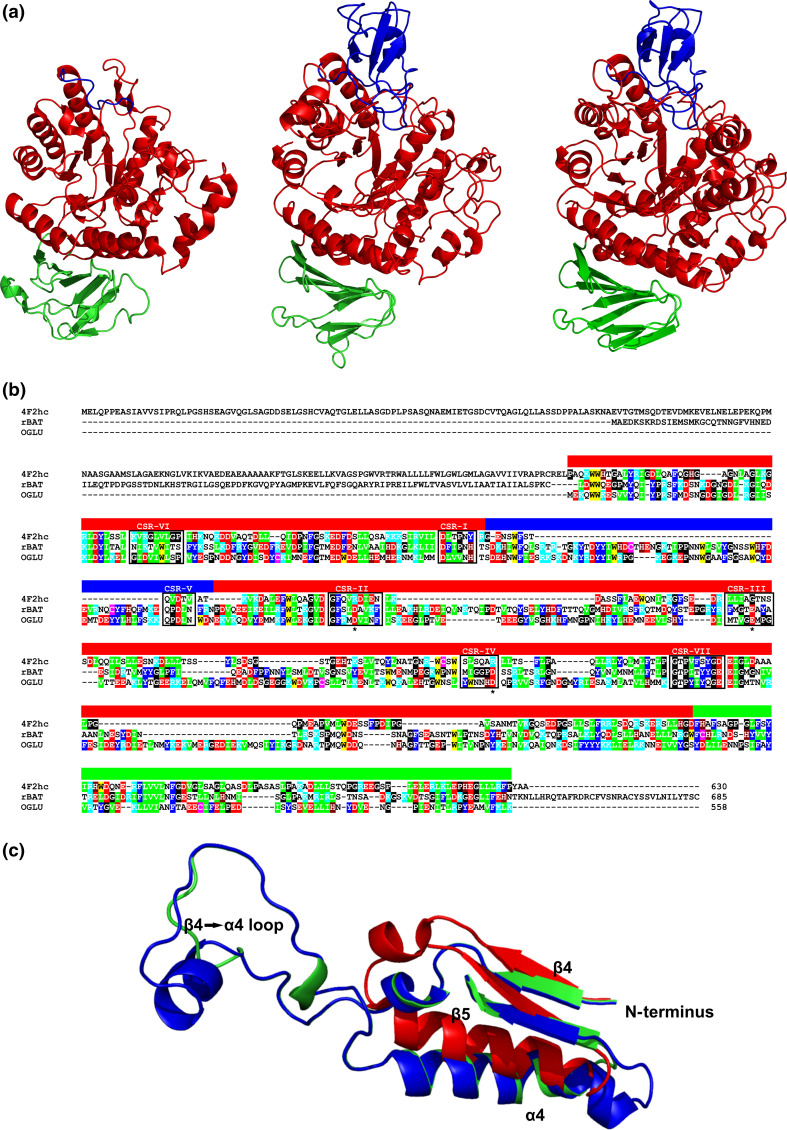
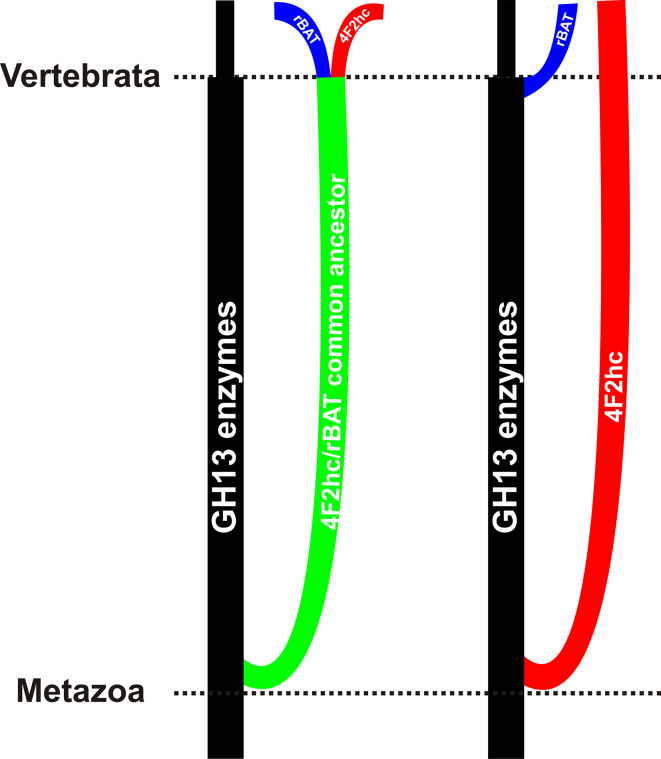
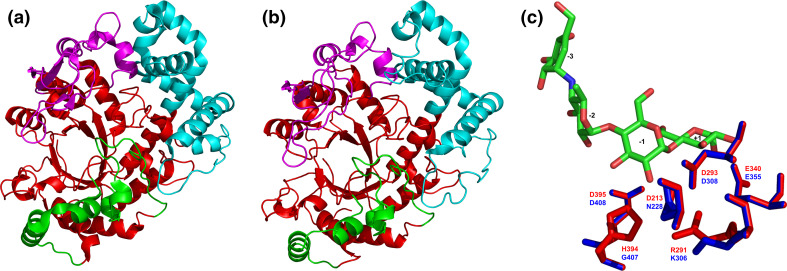
The α-amylase is a ubiquitous starch hydrolase catalyzing the cleavage of the α-1,4-glucosidic bonds in an endo-fashion. Various α-amylases originating from different taxonomic sources may differ from each other significantly in their exact substrate preference and product profile. Moreover, it also seems to be clear that at least two different amino acid sequences utilizing two different catalytic machineries have evolved to execute the same α-amylolytic specificity. The two have been classified in the Cabohydrate-Active enZyme database, the CAZy, in the glycoside hydrolase (GH) families GH13 and GH57. While the former and the larger α-amylase family GH13 evidently forms the clan GH-H with the families GH70 and GH77, the latter and the smaller α-amylase family GH57 has only been predicted to maybe define a future clan with the family GH119. Sequences and several tens of enzyme specificities found throughout all three kingdoms in many taxa provide an interesting material for evolutionarily oriented studies that have demonstrated remarkable observations. This review emphasizes just the three of them: (1) a close relatedness between the plant and archaeal α-amylases from the family GH13; (2) a common ancestry in the family GH13 of animal heavy chains of heteromeric amino acid transporter rBAT and 4F2 with the microbial α-glucosidases; and (3) the unique sequence features in the primary structures of amylomaltases from the genus Borrelia from the family GH77. Although the three examples cannot represent an exhaustive list of exceptional topics worth to be interested in, they may demonstrate the importance these enzymes possess in the overall scientific context.
Keywords: Evolutionary relatedness; Family GH77 amylomaltases of borrelian origin; Heavy-chains of rBAT and 4F2 proteins; Plant and archaeal α-amylases; α-Amylase family GH13.
Figures

References
-
- Søgaard M, Ji Abe, Martin-Eauclaire MF, Svensson B. α-Amylases: structure and function. Carbohydr Polym. 1993;21:137–146. doi: 10.1016/0144-8617(93)90008-R. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
