

Hepatitis B virus inhibits insulin receptor signaling and impairs liver regeneration via intracellular retention of the insulin receptor
- PMID: 27155659
- PMCID: PMC11108314
- DOI: 10.1007/s00018-016-2259-1
Hepatitis B virus inhibits insulin receptor signaling and impairs liver regeneration via intracellular retention of the insulin receptor
Abstract
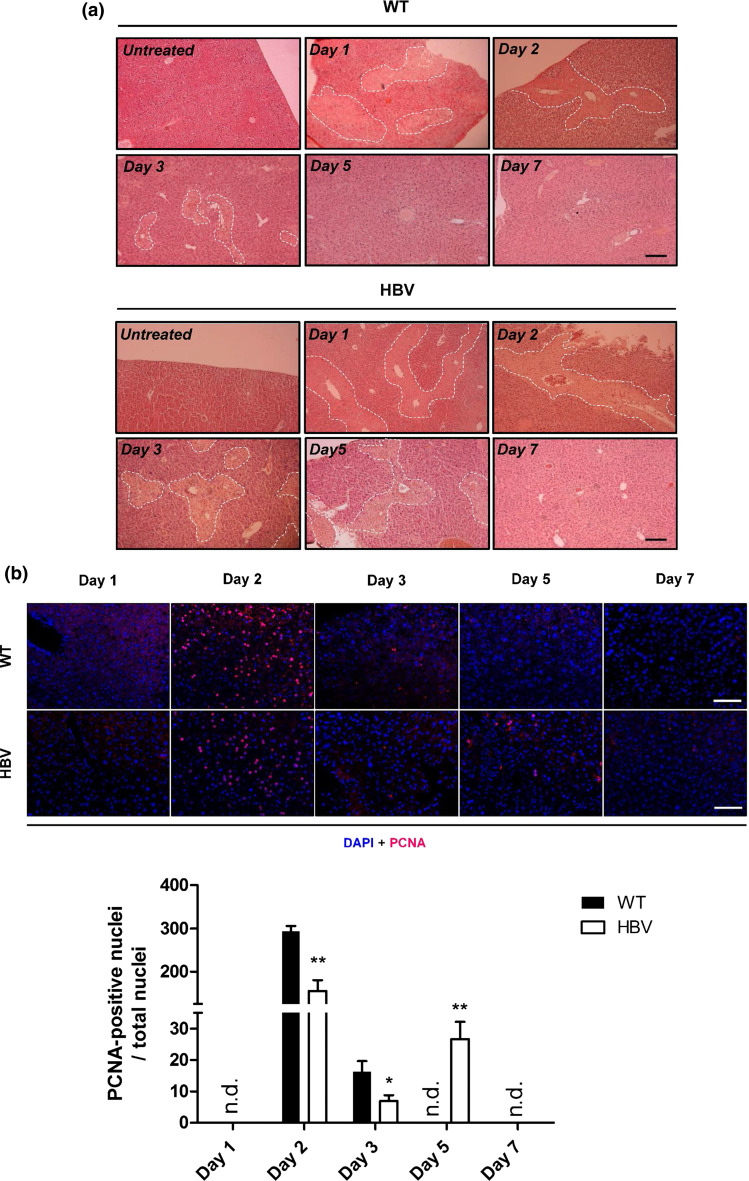
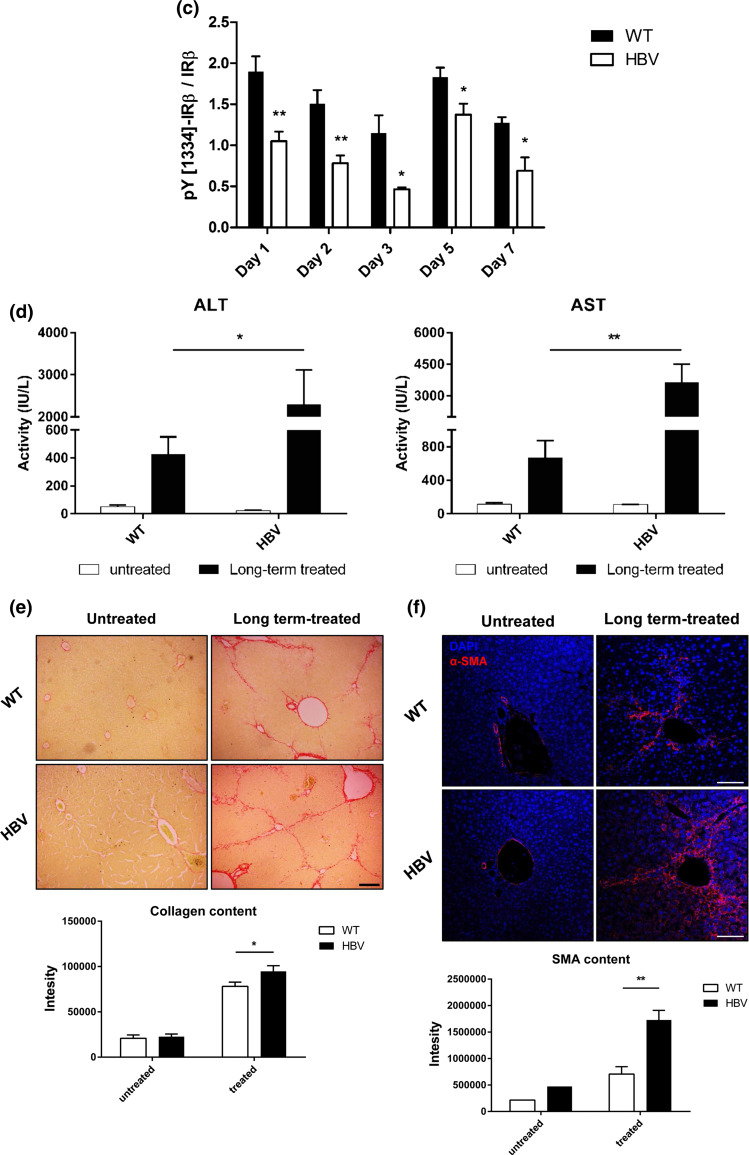
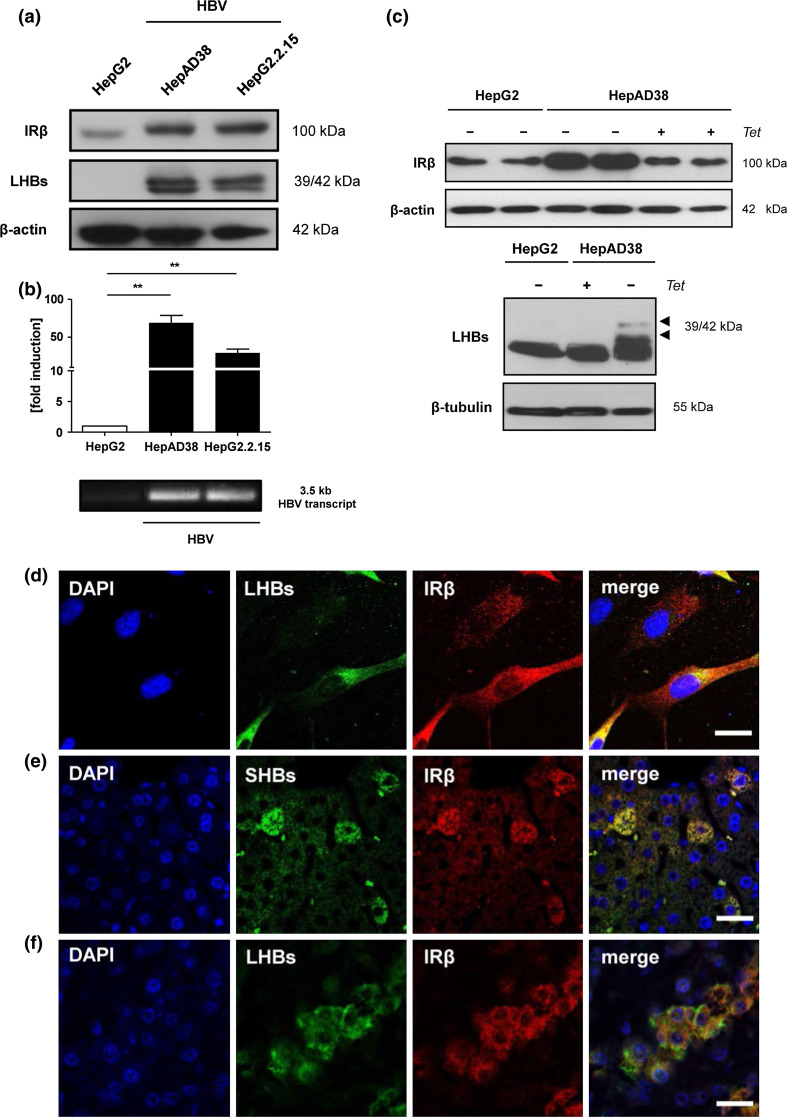
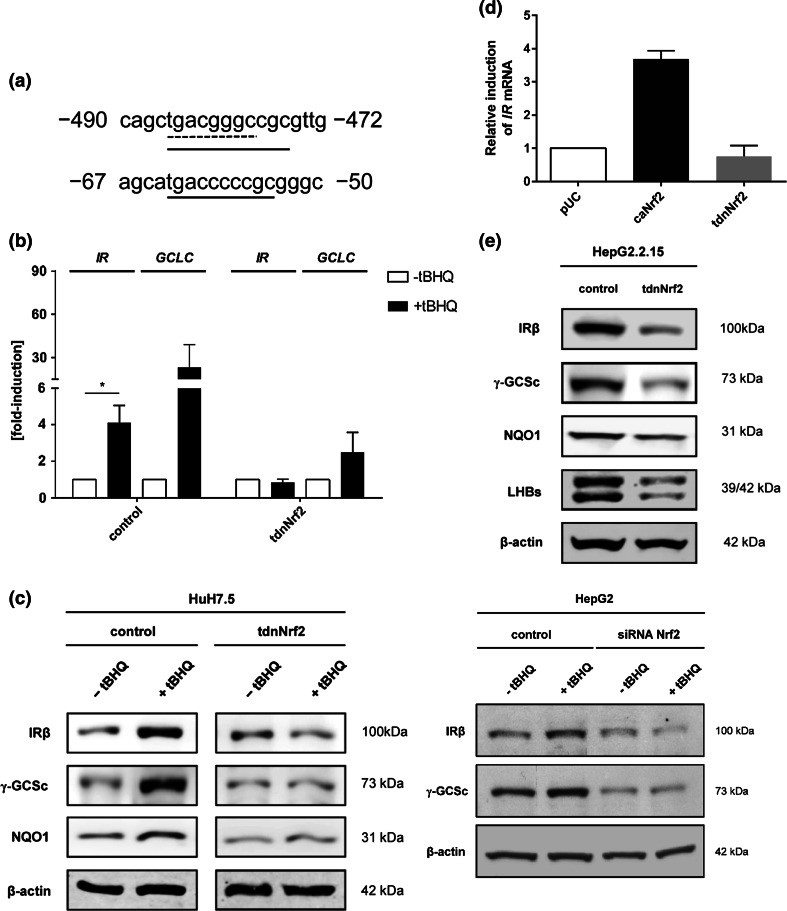
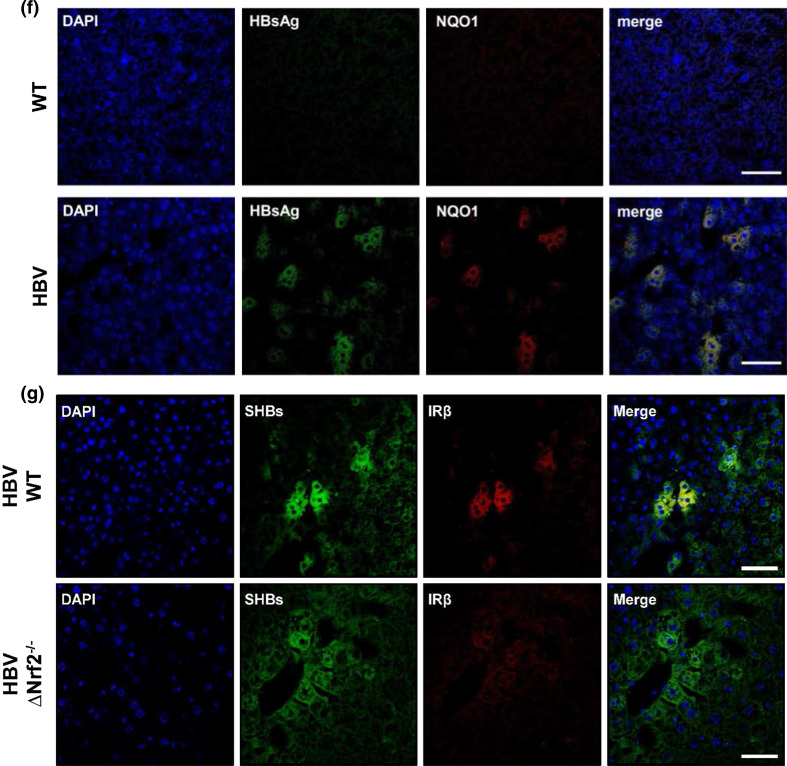
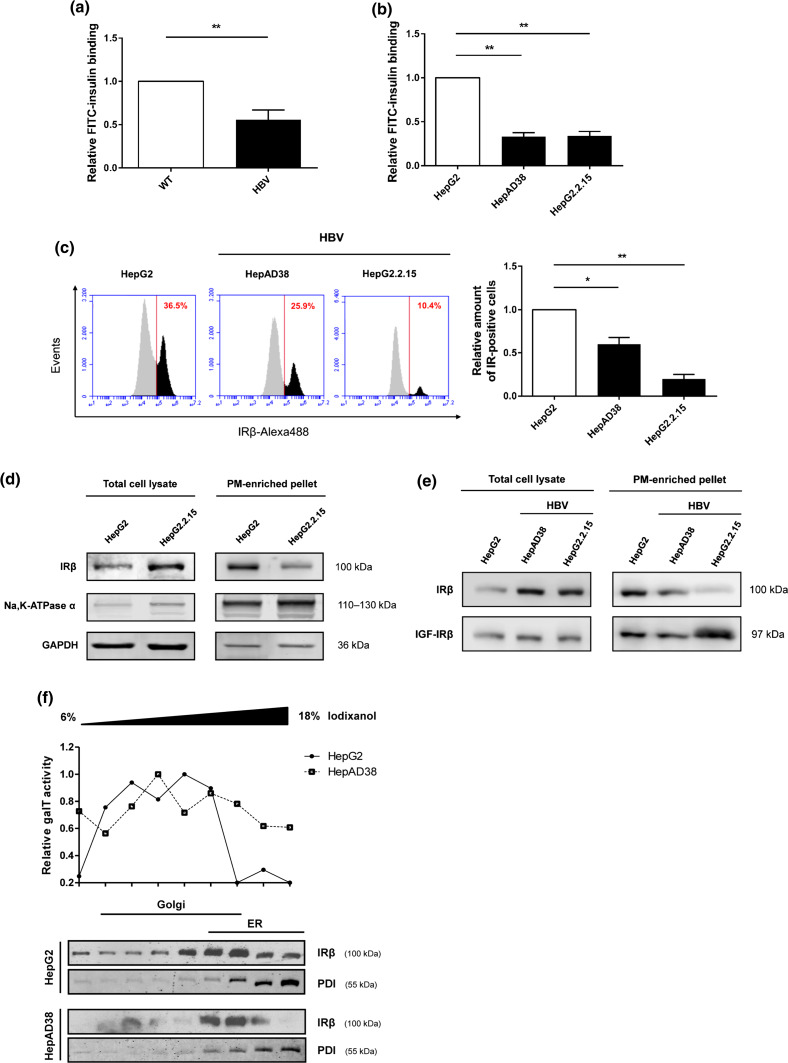
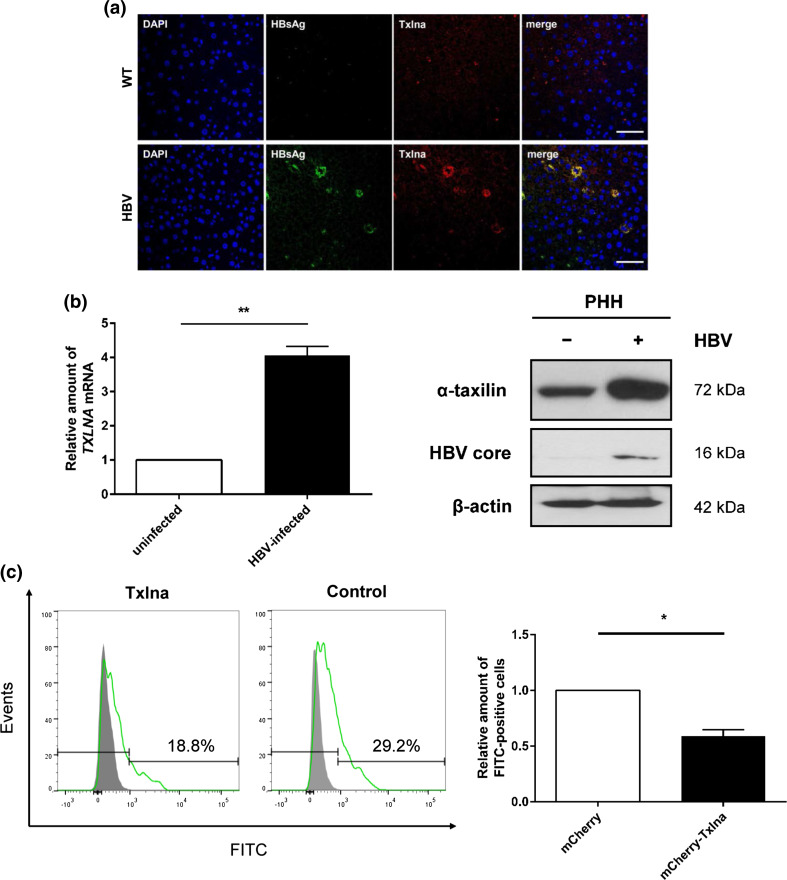
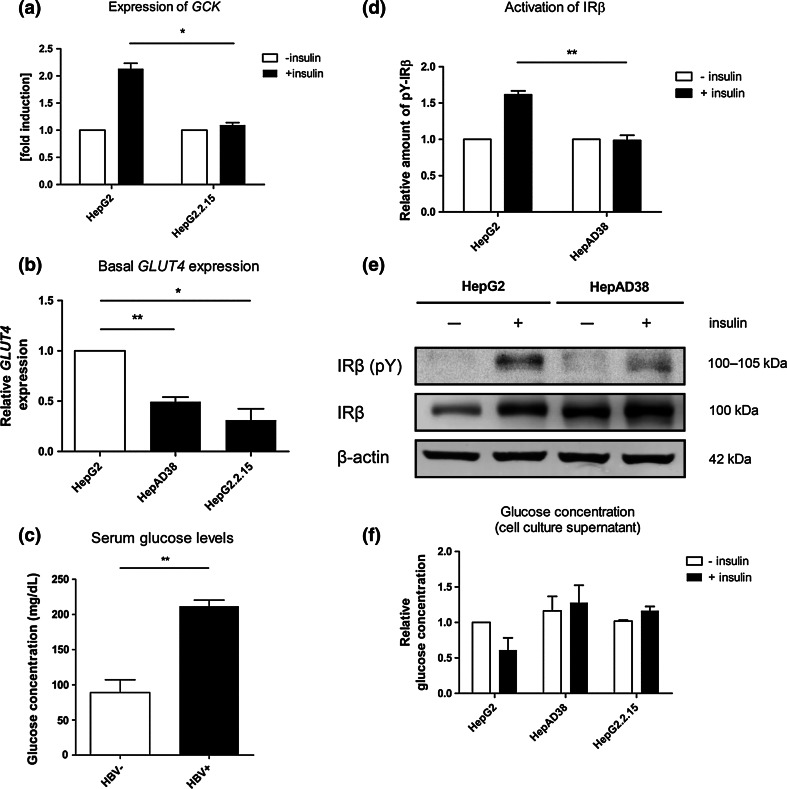
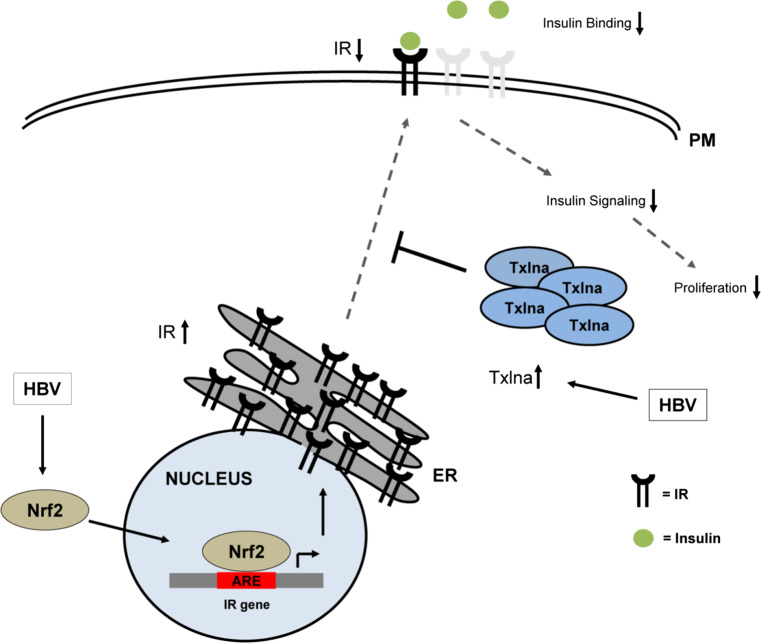
Hepatitis B virus (HBV) causes severe liver disease but the underlying mechanisms are incompletely understood. During chronic HBV infection, the liver is recurrently injured by immune cells in the quest for viral elimination. To compensate tissue injury, liver regeneration represents a vital process which requires proliferative insulin receptor signaling. This study aims to investigate the impact of HBV on liver regeneration and hepatic insulin receptor signaling. After carbon tetrachloride-induced liver injury, liver regeneration is delayed in HBV transgenic mice. These mice show diminished hepatocyte proliferation and increased expression of fibrosis markers. This is in accordance with a reduced activation of the insulin receptor although HBV induces expression of the insulin receptor via activation of NF-E2-related factor 2. This leads to increased intracellular amounts of insulin receptor in HBV expressing hepatocytes. However, intracellular retention of the receptor simultaneously reduces the amount of functional insulin receptors on the cell surface and thereby attenuates insulin binding in vitro and in vivo. Intracellular retention of the insulin receptor is caused by elevated amounts of α-taxilin, a free syntaxin binding protein, in HBV expressing hepatocytes preventing proper targeting of the insulin receptor to the cell surface. Consequently, functional analyses of insulin responsiveness revealed that HBV expressing hepatocytes are less sensitive to insulin stimulation leading to delayed liver regeneration. This study describes a novel pathomechanism that uncouples HBV expressing hepatocytes from proliferative signals and thereby impedes compensatory liver regeneration after liver injury.
Keywords: HBV; Insulin receptor signaling; Insulin resistance; Liver disease; Nrf2; α-Taxilin.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article.
Figures
Similar articles
-
Identification of α-taxilin as an essential factor for the life cycle of hepatitis B virus.J Hepatol. 2013 Nov;59(5):934-41. doi: 10.1016/j.jhep.2013.06.020. Epub 2013 Jun 28. J Hepatol. 2013. PMID: 23816704
-
Viral-load-dependent effects of liver injury and regeneration on hepatitis B virus replication in mice.J Virol. 2012 Sep;86(18):9599-605. doi: 10.1128/JVI.01087-12. Epub 2012 Jun 20. J Virol. 2012. PMID: 22718822 Free PMC article.
-
Hepatitis B virus inhibits liver regeneration via epigenetic regulation of urokinase-type plasminogen activator.Hepatology. 2013 Aug;58(2):762-76. doi: 10.1002/hep.26379. Epub 2013 Jun 24. Hepatology. 2013. PMID: 23483589
-
The cytoprotective Nrf2 transcription factor controls insulin receptor signaling in the regenerating liver.Cell Cycle. 2008 Apr 1;7(7):874-8. doi: 10.4161/cc.7.7.5617. Epub 2008 Jan 18. Cell Cycle. 2008. PMID: 18414027 Review.
-
Hepatitis B Virus DNA Integration and Clonal Expansion of Hepatocytes in the Chronically Infected Liver.Viruses. 2021 Jan 30;13(2):210. doi: 10.3390/v13020210. Viruses. 2021. PMID: 33573130 Free PMC article. Review.
Cited by
-
Therapeutic Modulation of Virus-Induced Oxidative Stress via the Nrf2-Dependent Antioxidative Pathway.Oxid Med Cell Longev. 2018 Oct 31;2018:6208067. doi: 10.1155/2018/6208067. eCollection 2018. Oxid Med Cell Longev. 2018. PMID: 30515256 Free PMC article. Review.
-
Hepatitis B Virus Infection and Increased Risk of Gestational Diabetes Regardless of Liver Function Status: A Xiamen Area Population-Based Study.Front Physiol. 2022 Jul 11;13:938149. doi: 10.3389/fphys.2022.938149. eCollection 2022. Front Physiol. 2022. PMID: 35899024 Free PMC article.
-
Influencing factors and mechanism of hepatocyte regeneration.J Transl Med. 2025 Apr 30;23(1):493. doi: 10.1186/s12967-025-06278-9. J Transl Med. 2025. PMID: 40307789 Free PMC article. Review.
-
Chronic Viral Hepatitis Signifies the Association of Premixed Insulin Analogues with Liver Cancer Risks: A Nationwide Population-Based Study.Int J Environ Res Public Health. 2019 Jun 13;16(12):2097. doi: 10.3390/ijerph16122097. Int J Environ Res Public Health. 2019. PMID: 31200528 Free PMC article.
-
Diabetogenic viruses: linking viruses to diabetes mellitus.Heliyon. 2023 Mar 30;9(4):e15021. doi: 10.1016/j.heliyon.2023.e15021. eCollection 2023 Apr. Heliyon. 2023. PMID: 37064445 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
