

Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas
- PMID: 27158780
- PMCID: PMC4884143
- DOI: 10.1038/ng.3564
Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas
Abstract
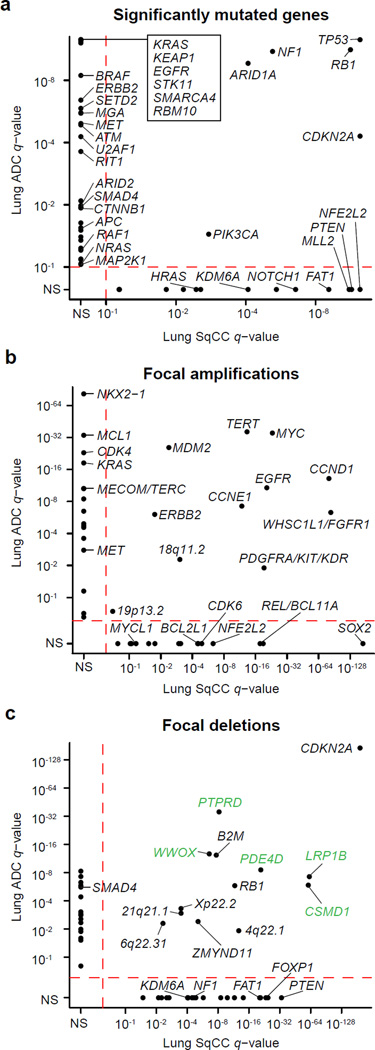
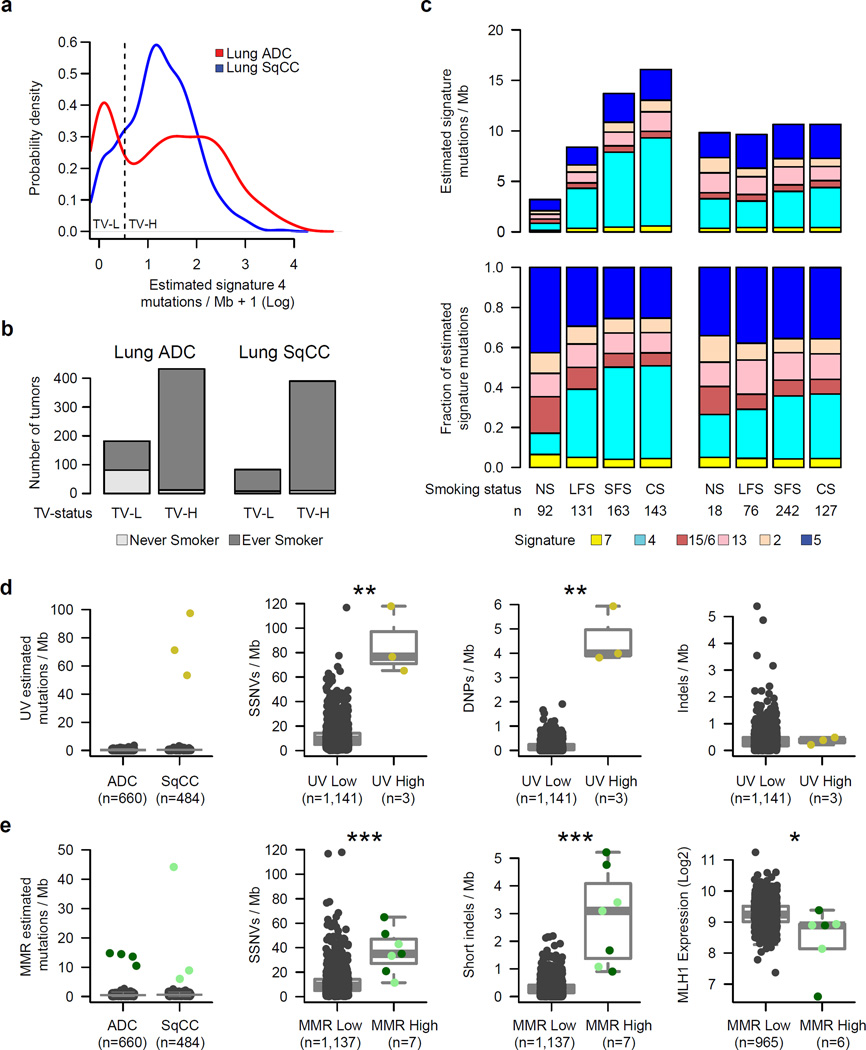
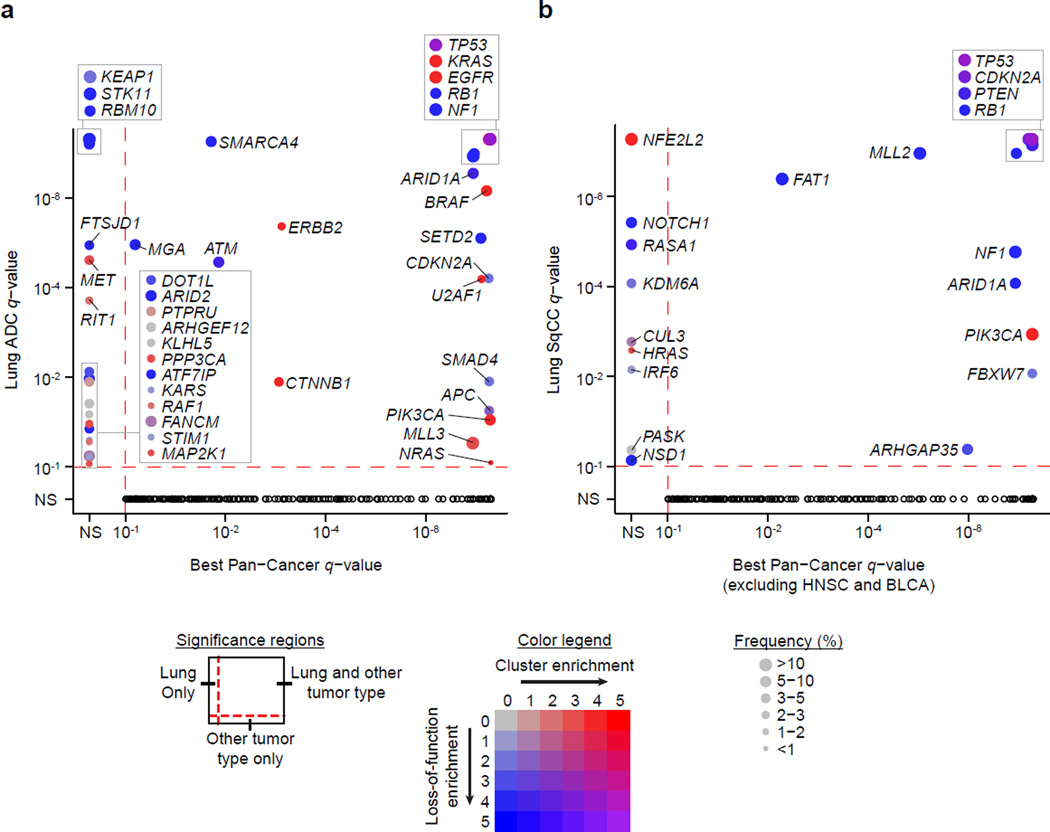
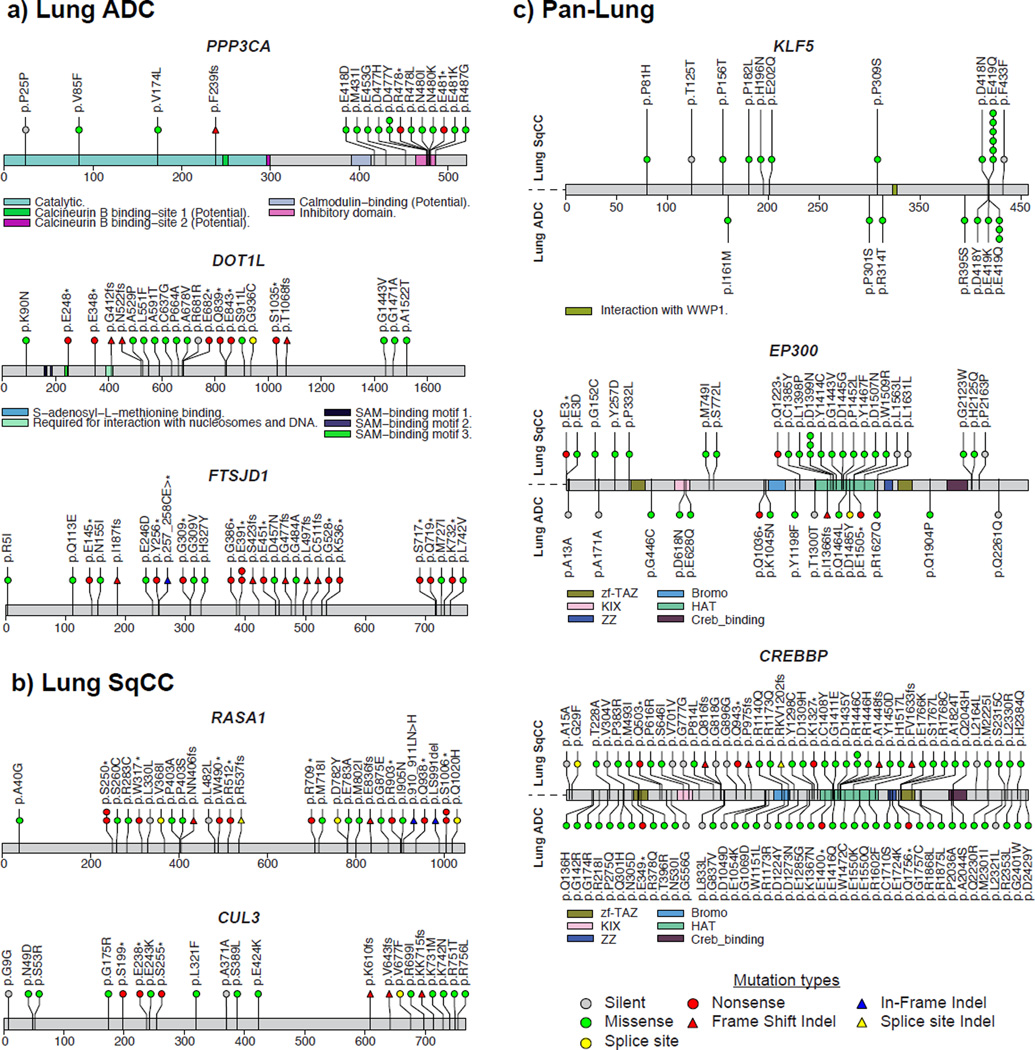
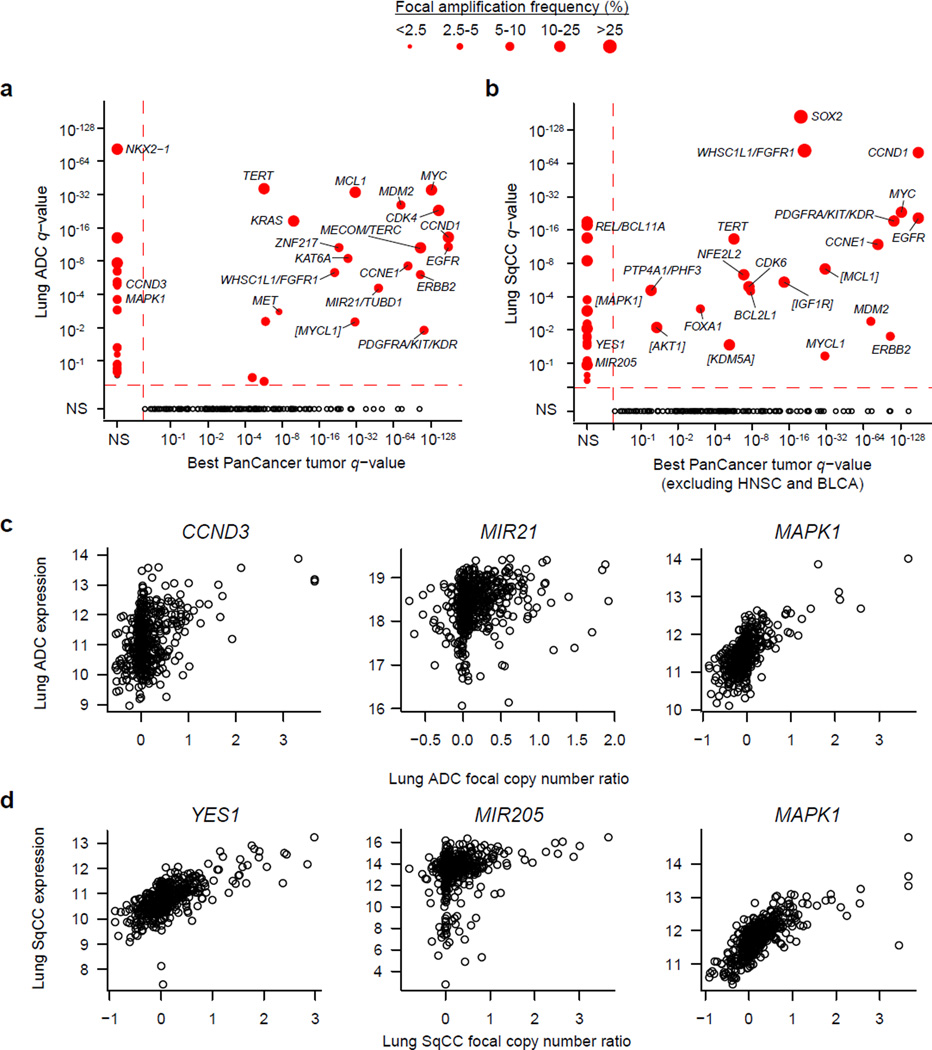
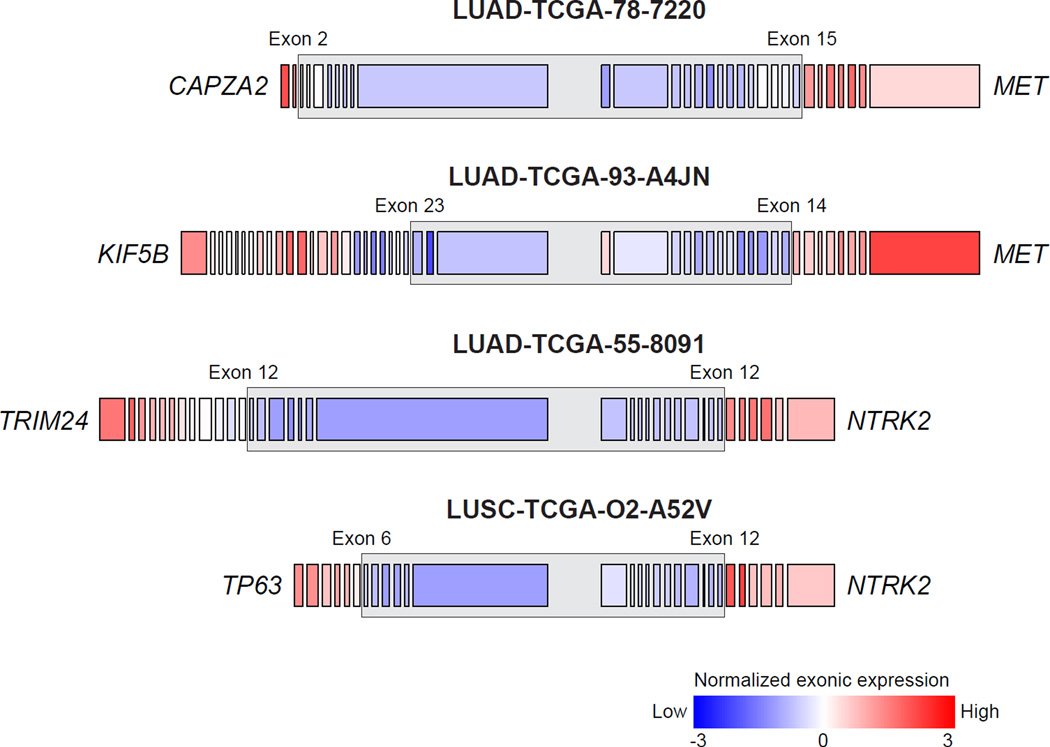
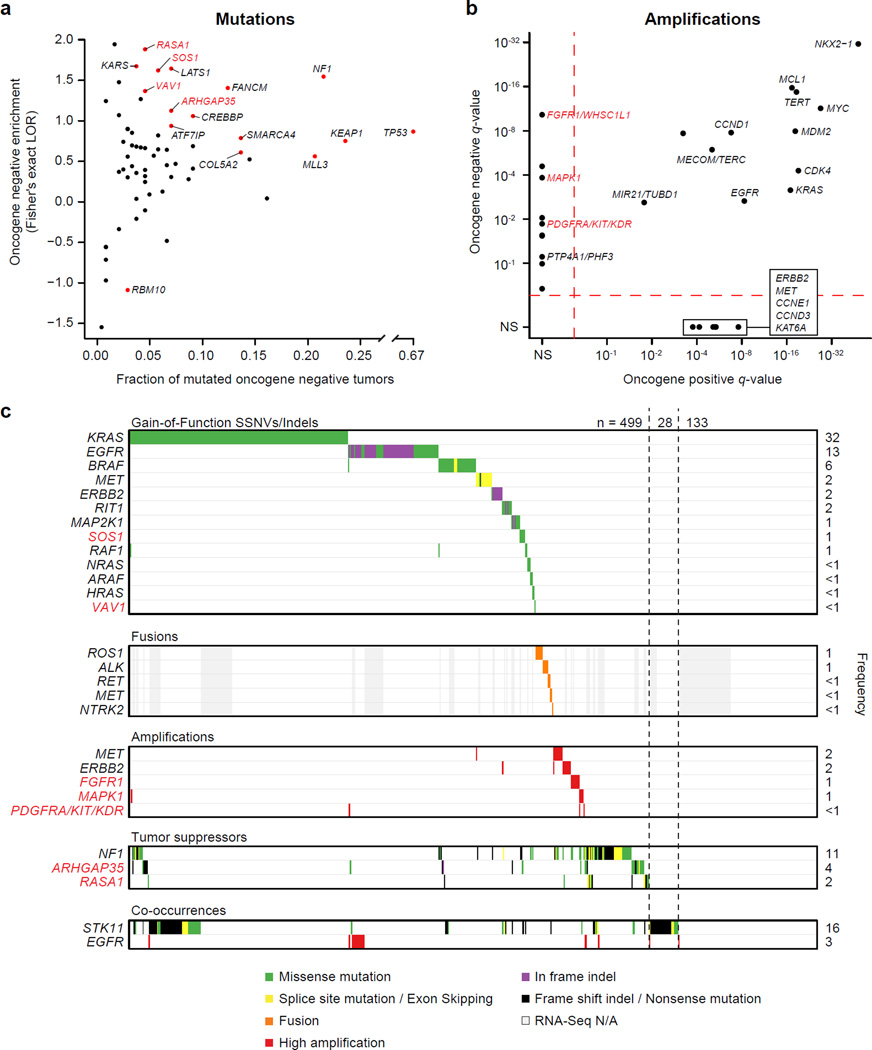
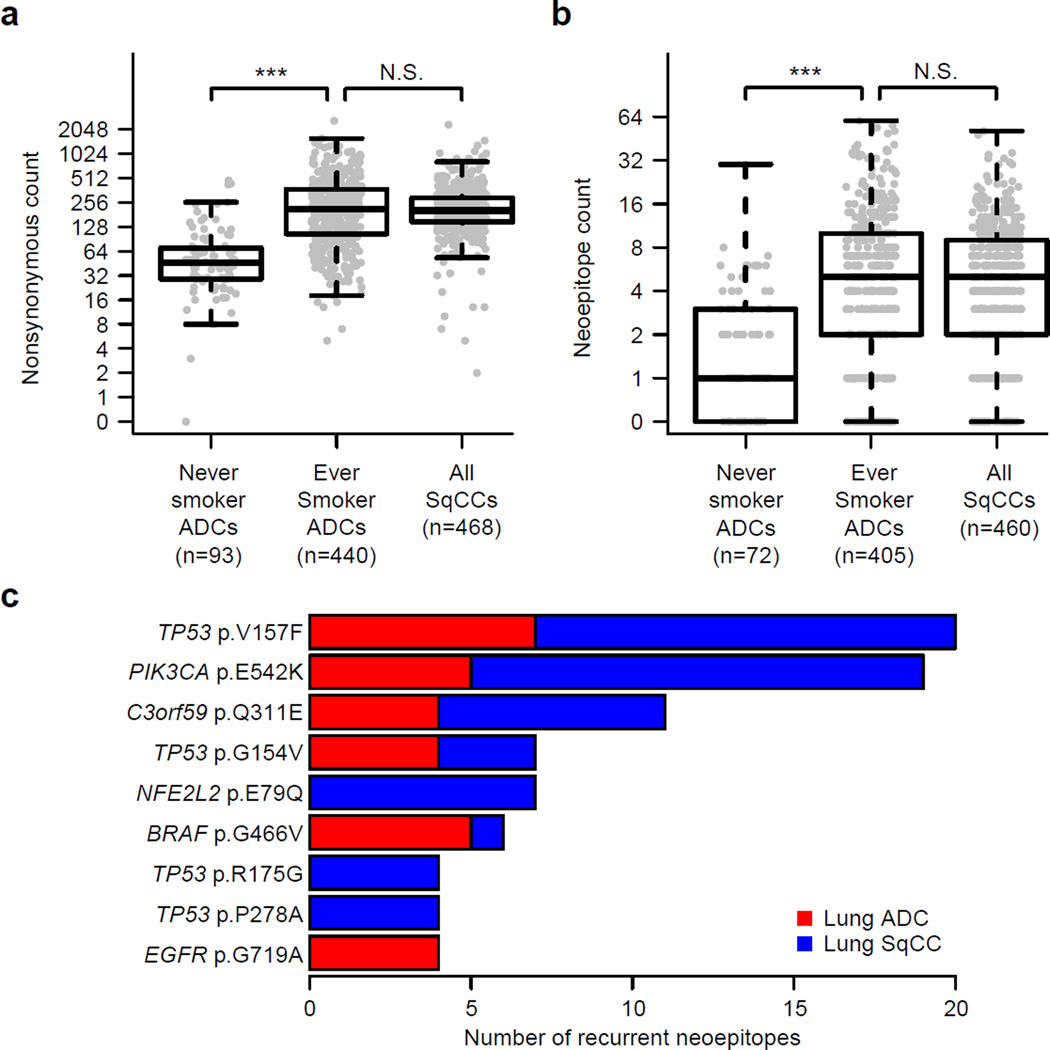
To compare lung adenocarcinoma (ADC) and lung squamous cell carcinoma (SqCC) and to identify new drivers of lung carcinogenesis, we examined the exome sequences and copy number profiles of 660 lung ADC and 484 lung SqCC tumor-normal pairs. Recurrent alterations in lung SqCCs were more similar to those of other squamous carcinomas than to alterations in lung ADCs. New significantly mutated genes included PPP3CA, DOT1L, and FTSJD1 in lung ADC, RASA1 in lung SqCC, and KLF5, EP300, and CREBBP in both tumor types. New amplification peaks encompassed MIR21 in lung ADC, MIR205 in lung SqCC, and MAPK1 in both. Lung ADCs lacking receptor tyrosine kinase-Ras-Raf pathway alterations had mutations in SOS1, VAV1, RASA1, and ARHGAP35. Regarding neoantigens, 47% of the lung ADC and 53% of the lung SqCC tumors had at least five predicted neoepitopes. Although targeted therapies for lung ADC and SqCC are largely distinct, immunotherapies may aid in treatment for both subtypes.
Figures
Comment in
-
Next-generation molecular therapy in lung cancer.Transl Lung Cancer Res. 2018 Feb;7(Suppl 1):S31-S34. doi: 10.21037/tlcr.2018.01.03. Transl Lung Cancer Res. 2018. PMID: 29531901 Free PMC article. No abstract available.
References
Methods-only references
-
- Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P30 CA016672/CA/NCI NIH HHS/United States
- K99 CA197762/CA/NCI NIH HHS/United States
- K08 CA163677/CA/NCI NIH HHS/United States
- T32 HG002295/HG/NHGRI NIH HHS/United States
- U24 CA144025/CA/NCI NIH HHS/United States
- R35 CA197568/CA/NCI NIH HHS/United States
- U24 CA143883/CA/NCI NIH HHS/United States
- U24 CA126546/CA/NCI NIH HHS/United States
- U24 CA126544/CA/NCI NIH HHS/United States
- P01 CA120964/CA/NCI NIH HHS/United States
- U24 CA143845/CA/NCI NIH HHS/United States
- R01 CA184922/CA/NCI NIH HHS/United States
- R01 CA155010/CA/NCI NIH HHS/United States
- U24 CA143848/CA/NCI NIH HHS/United States
- U24 CA143867/CA/NCI NIH HHS/United States
- U24 CA199461/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
