

Cladribine and Fludarabine Nucleotides Induce Distinct Hexamers Defining a Common Mode of Reversible RNR Inhibition
- PMID: 27159113
- PMCID: PMC4946967
- DOI: 10.1021/acschembio.6b00303
Cladribine and Fludarabine Nucleotides Induce Distinct Hexamers Defining a Common Mode of Reversible RNR Inhibition
Abstract
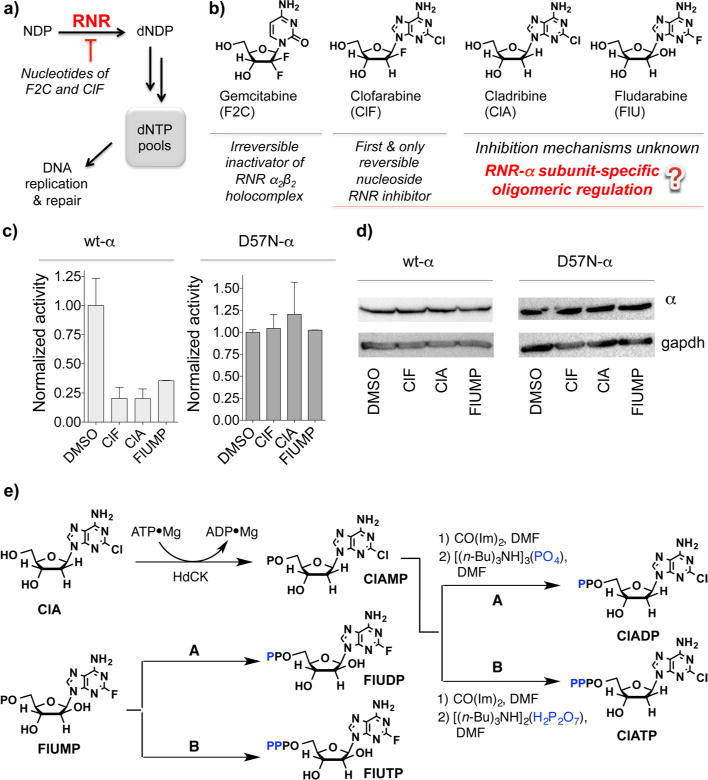
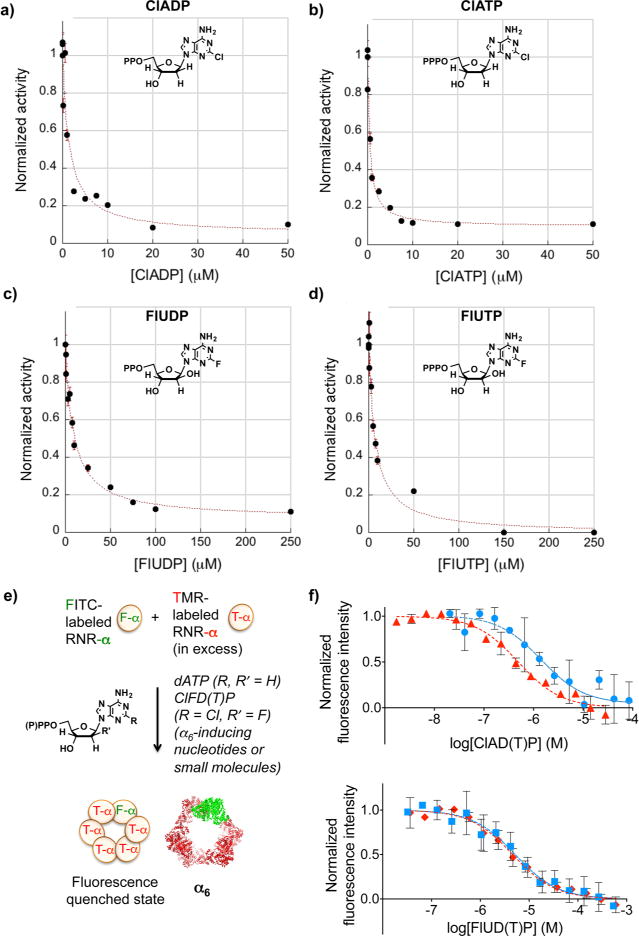
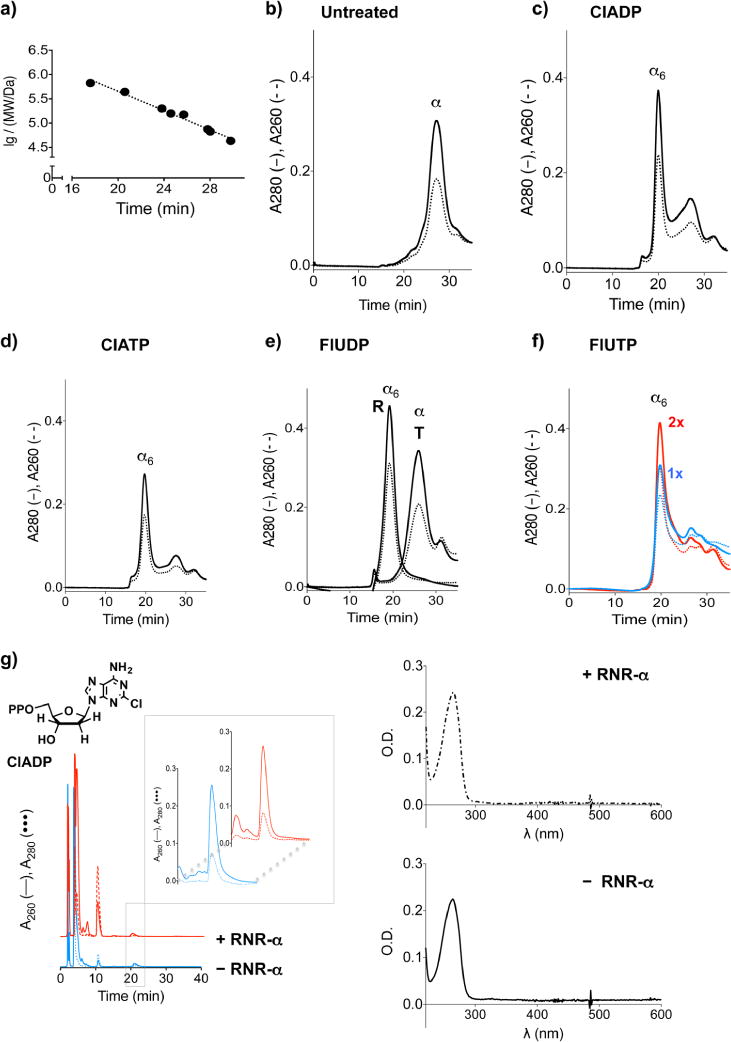
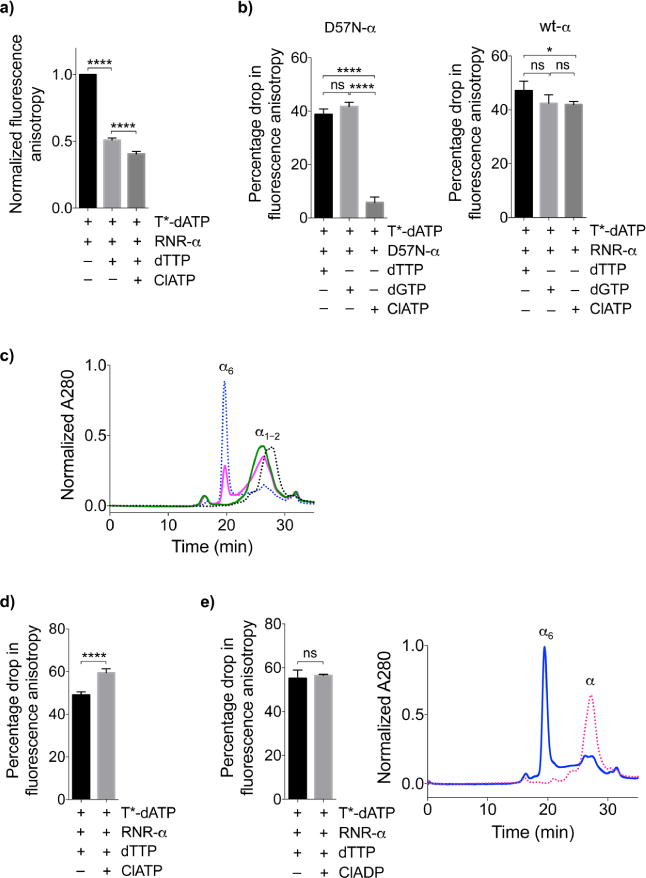
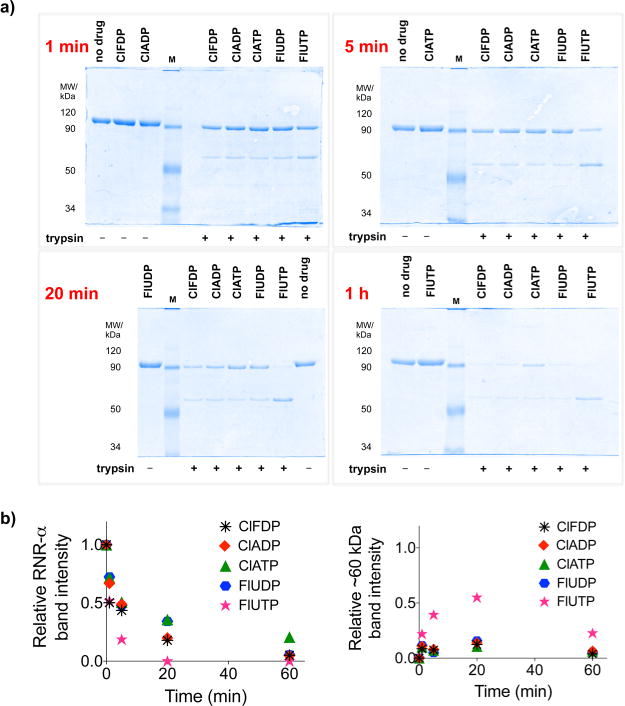
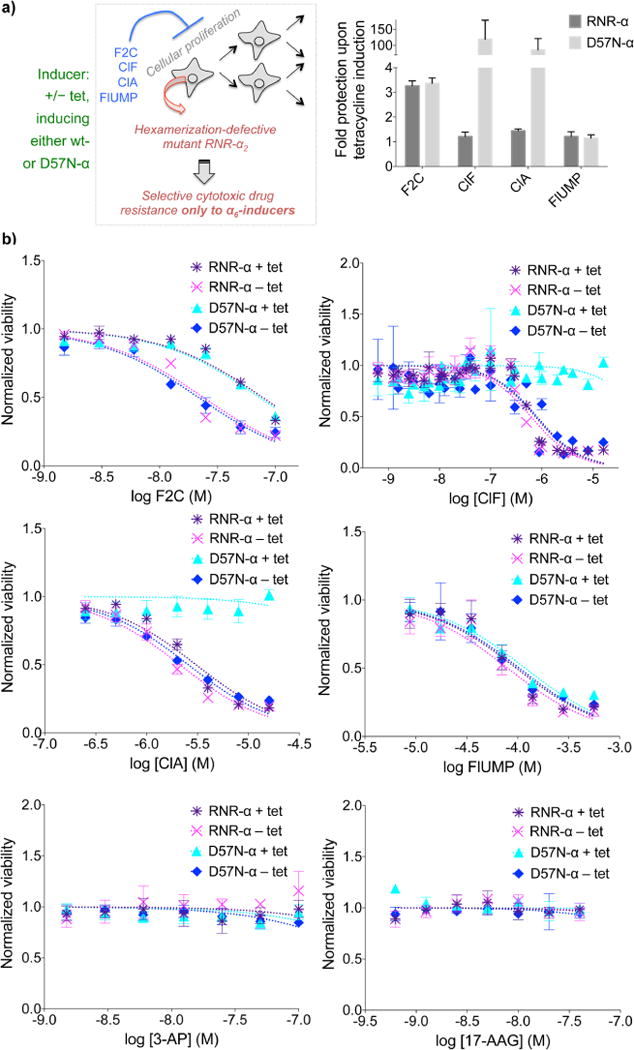
The enzyme ribonucleotide reductase (RNR) is a major target of anticancer drugs. Until recently, suicide inactivation in which synthetic substrate analogs (nucleoside diphosphates) irreversibly inactivate the RNR-α2β2 heterodimeric complex was the only clinically proven inhibition pathway. For instance, this mechanism is deployed by the multifactorial anticancer agent gemcitabine diphosphate. Recently reversible targeting of RNR-α-alone coupled with ligand-induced RNR-α-persistent hexamerization has emerged to be of clinical significance. To date, clofarabine nucleotides are the only known example of this mechanism. Herein, chemoenzymatic syntheses of the active forms of two other drugs, phosphorylated cladribine (ClA) and fludarabine (FlU), allow us to establish that reversible inhibition is common to numerous drugs in clinical use. Enzyme inhibition and fluorescence anisotropy assays show that the di- and triphosphates of the two nucleosides function as reversible (i.e., nonmechanism-based) inhibitors of RNR and interact with the catalytic (C site) and the allosteric activity (A site) sites of RNR-α, respectively. Gel filtration, protease digestion, and FRET assays demonstrate that inhibition is coupled with formation of conformationally diverse hexamers. Studies in 293T cells capable of selectively inducing either wild-type or oligomerization-defective mutant RNR-α overexpression delineate the central role of RNR-α oligomerization in drug activity, and highlight a potential resistance mechanism to these drugs. These data set the stage for new interventions targeting RNR oligomeric regulation.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
-
- Terstappen GC, Schlupen C, Raggiaschi R, Gaviraghi G. Target deconvolution strategies in drug discovery. Nat Rev Drug Discovery. 2007;6:891–903. - PubMed
-
- Stubbe J, van der Donk WA. Ribonucleotide reductases: radical enzymes with suicidal tendencies. Chem Biol. 1995;2:793–801. - PubMed
-
- Nordlund P, Reichard P. Ribonucleotide reductases. Annu Rev Biochem. 2006;75:681–706. - PubMed
-
- Aye Y, Li M, Long MJ, Weiss RS. Ribonucleotide reductase and cancer: biological mechanisms and targeted therapies. Oncogene. 2015;34:2011–2021. - PubMed
-
- Licht S, Stubbe J. In: Mechanistic Investigations of Ribonucleotide Reductases. Poulter CD, editor. Elsevier; Amsterdam: 1999. p. 163.p. 203.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
