

Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis
- PMID: 27159577
- PMCID: PMC4920070
- DOI: 10.1038/nchembio.2079
Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis
Abstract
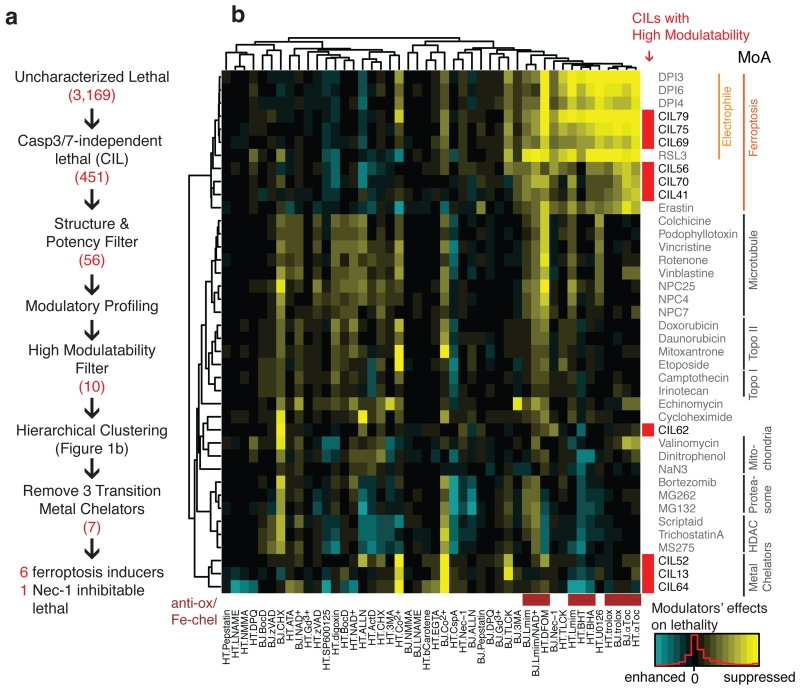
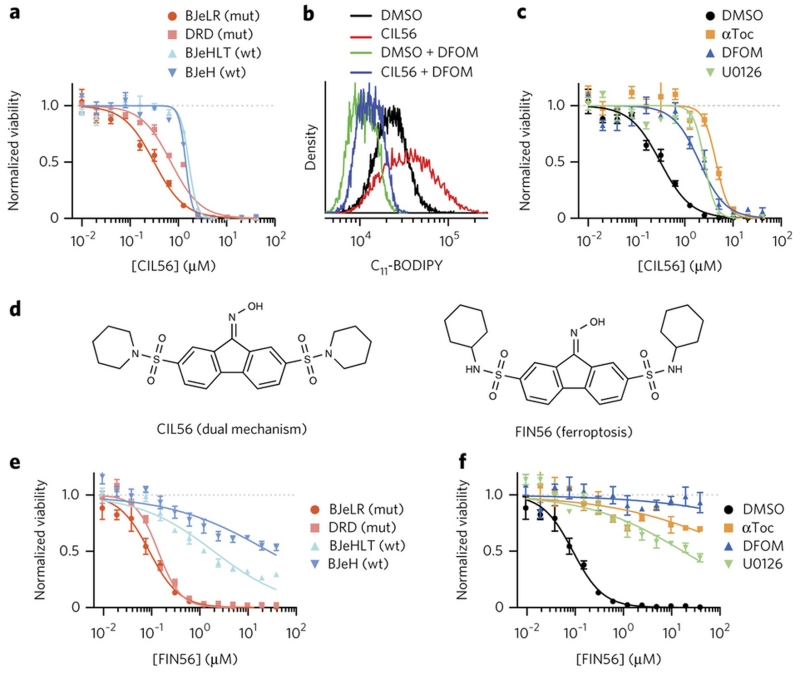
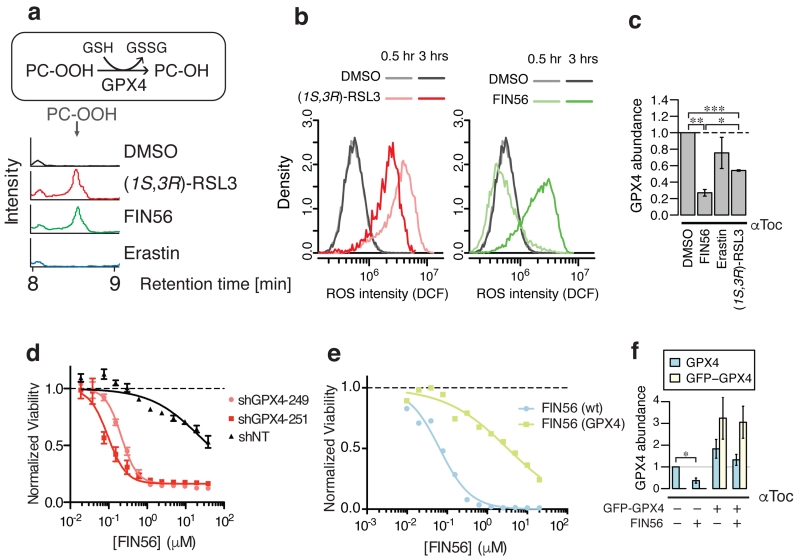
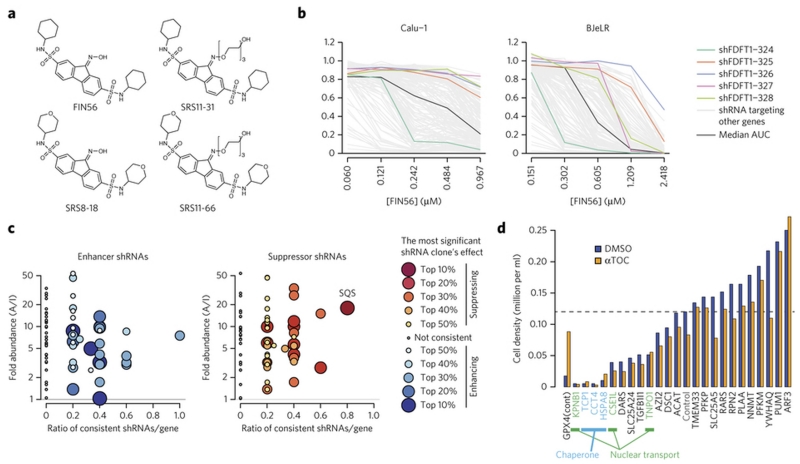
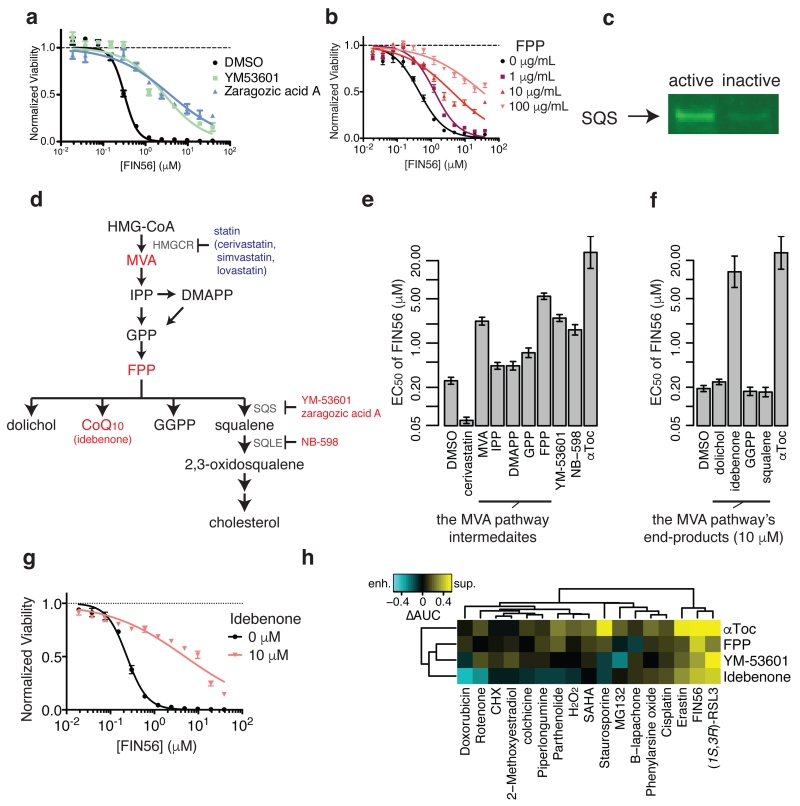
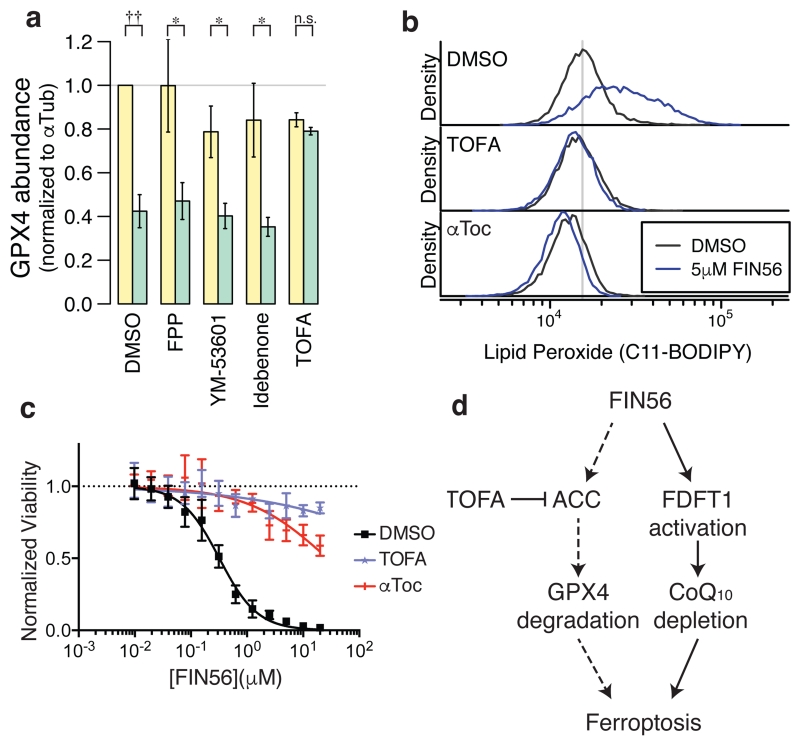
Apoptosis is one type of programmed cell death. Increasingly, non-apoptotic cell death is recognized as being genetically controlled, or 'regulated'. However, the full extent and diversity of alternative cell death mechanisms remain uncharted. Here we surveyed the landscape of pharmacologically accessible cell death mechanisms. In an examination of 56 caspase-independent lethal compounds, modulatory profiling showed that 10 compounds induced three different types of regulated non-apoptotic cell death. Optimization of one of those ten resulted in the discovery of FIN56, a specific inducer of ferroptosis. Ferroptosis has been found to occur when the lipid-repair enzyme GPX4 is inhibited. FIN56 promoted degradation of GPX4. FIN56 also bound to and activated squalene synthase, an enzyme involved in isoprenoid biosynthesis, independent of GPX4 degradation. These discoveries show that dysregulation of lipid metabolism is associated with ferroptosis. This systematic approach is a means to discover and characterize novel cell death phenotypes.
Figures
Comment in
-
Chemical genetics: Unraveling cell death mysteries.Nat Chem Biol. 2016 Jun 17;12(7):470-1. doi: 10.1038/nchembio.2110. Nat Chem Biol. 2016. PMID: 27315536 Free PMC article.
References
-
- Aravind L, Dixit VM, Koonin EV. The domains of death: evolution of the apoptosis machinery. Trends Biochem. Sci. 1999;24:47–53. - PubMed
-
- Berghe TV, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014;15:135–147. - PubMed
-
- Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: The Release of Damage-Associated Molecular Patterns and Its Physiological Relevance. Immunity. 2013;38:209–223. - PubMed
Methods-only references
-
- Cholody WM, et al. Derivatives of fluorene, anthracene, xanthene, dibenzosuberone and acridine and uses thereof. 2008 at < http://www.google.com/patents/WO2008140792A1>.
Publication types
MeSH terms
Substances
Associated data
- PubChem-Substance/313041240
- PubChem-Substance/313041241
- PubChem-Substance/313041242
- PubChem-Substance/313041243
- PubChem-Substance/313041244
- PubChem-Substance/313041245
- PubChem-Substance/313041246
- PubChem-Substance/313041247
- PubChem-Substance/313041248
- PubChem-Substance/313041249
- PubChem-Substance/313041250
- PubChem-Substance/313041251
- PubChem-Substance/313041252
- PubChem-Substance/313041253
- PubChem-Substance/313041254
- PubChem-Substance/313041255
- PubChem-Substance/313041256
- PubChem-Substance/313041257
- PubChem-Substance/313041258
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
