

Antibiotic resistance mechanisms in M. tuberculosis: an update
- PMID: 27161440
- PMCID: PMC4988520
- DOI: 10.1007/s00204-016-1727-6
Antibiotic resistance mechanisms in M. tuberculosis: an update
Abstract
Treatment of tuberculosis (TB) has been a therapeutic challenge because of not only the naturally high resistance level of Mycobacterium tuberculosis to antibiotics but also the newly acquired mutations that confer further resistance. Currently standardized regimens require patients to daily ingest up to four drugs under direct observation of a healthcare worker for a period of 6-9 months. Although they are quite effective in treating drug susceptible TB, these lengthy treatments often lead to patient non-adherence, which catalyzes for the emergence of M. tuberculosis strains that are increasingly resistant to the few available anti-TB drugs. The rapid evolution of M. tuberculosis, from mono-drug-resistant to multiple drug-resistant, extensively drug-resistant and most recently totally drug-resistant strains, is threatening to make TB once again an untreatable disease if new therapeutic options do not soon become available. Here, I discuss the molecular mechanisms by which M. tuberculosis confers its profound resistance to antibiotics. This knowledge may help in developing novel strategies for weakening drug resistance, thus enhancing the potency of available antibiotics against both drug susceptible and resistant M. tuberculosis strains.
Keywords: Antibiotic; Bactericidal; Drug resistance; Mechanism; Mycobacterium; Tuberculosis.
Figures
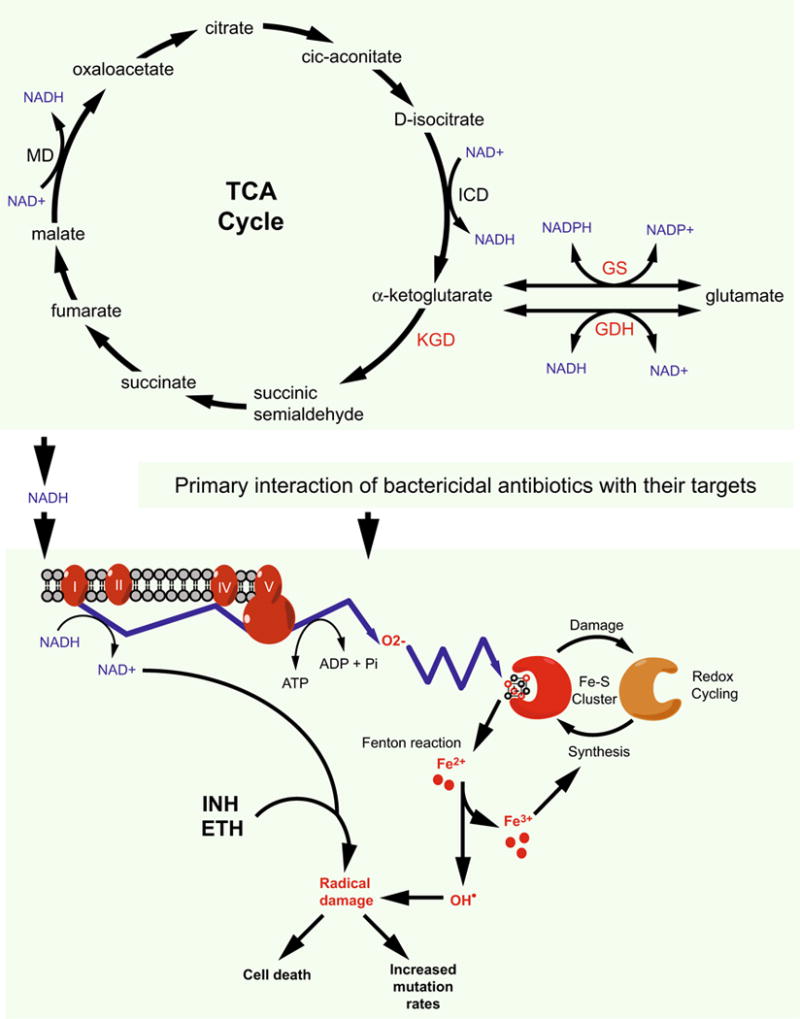
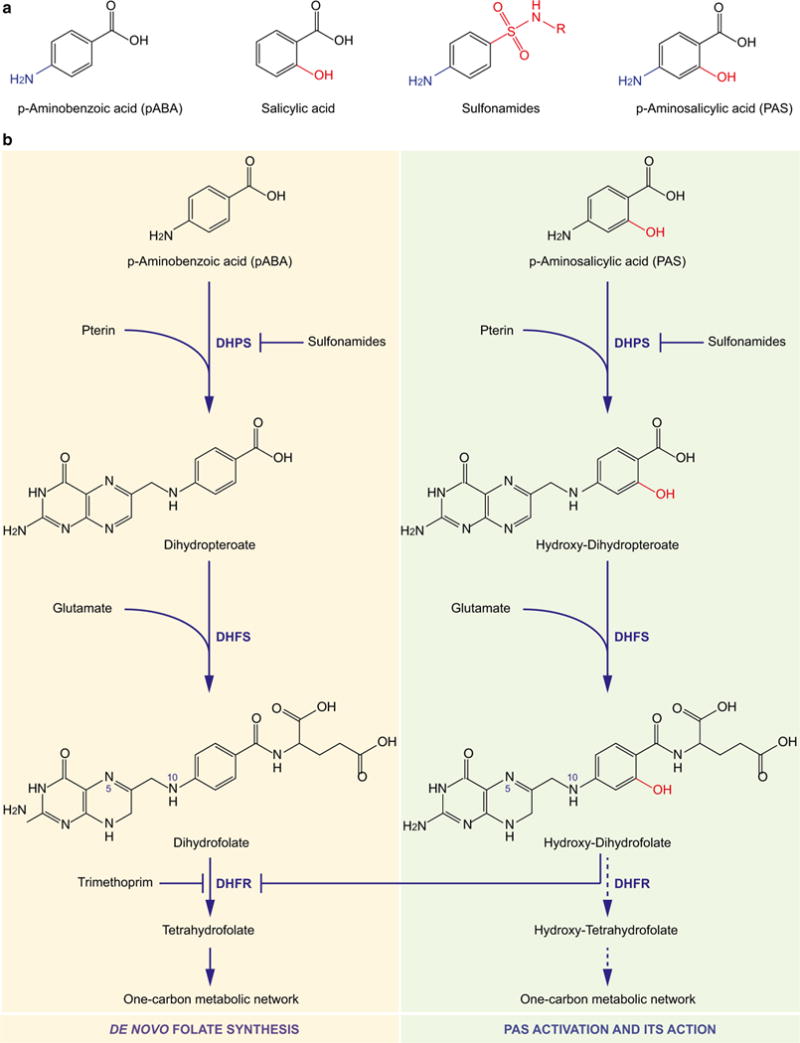
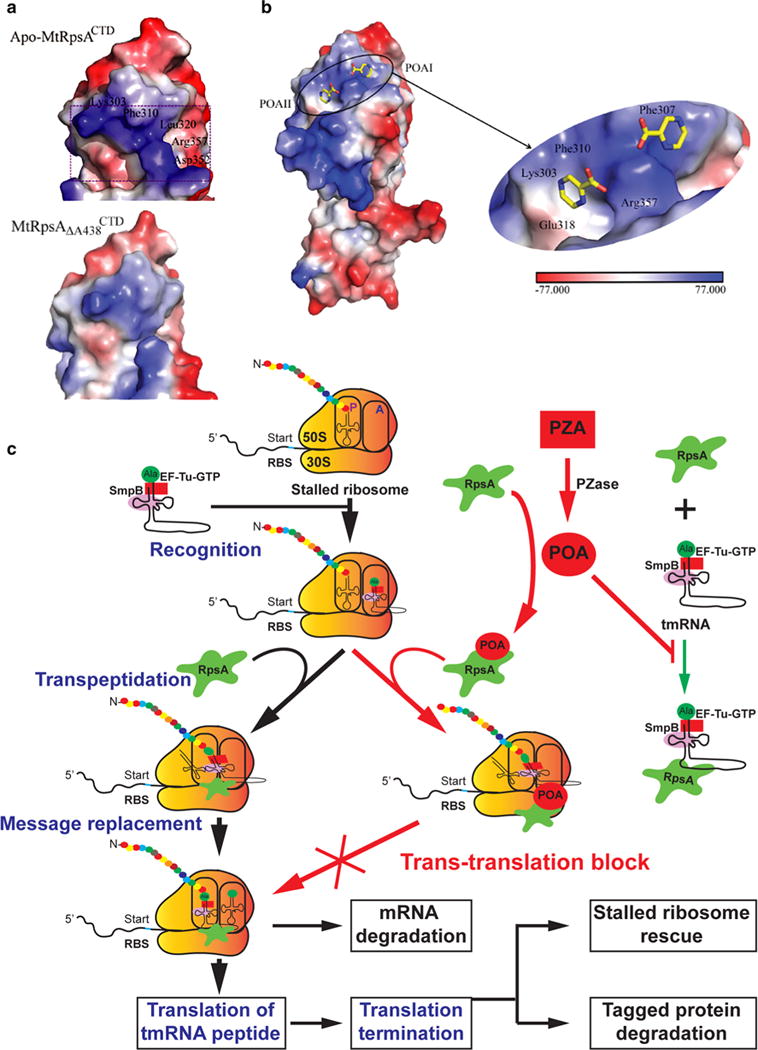
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
