

A Stochastic Point Cloud Sampling Method for Multi-Template Protein Comparative Modeling
- PMID: 27161489
- PMCID: PMC4861977
- DOI: 10.1038/srep25687
A Stochastic Point Cloud Sampling Method for Multi-Template Protein Comparative Modeling
Abstract
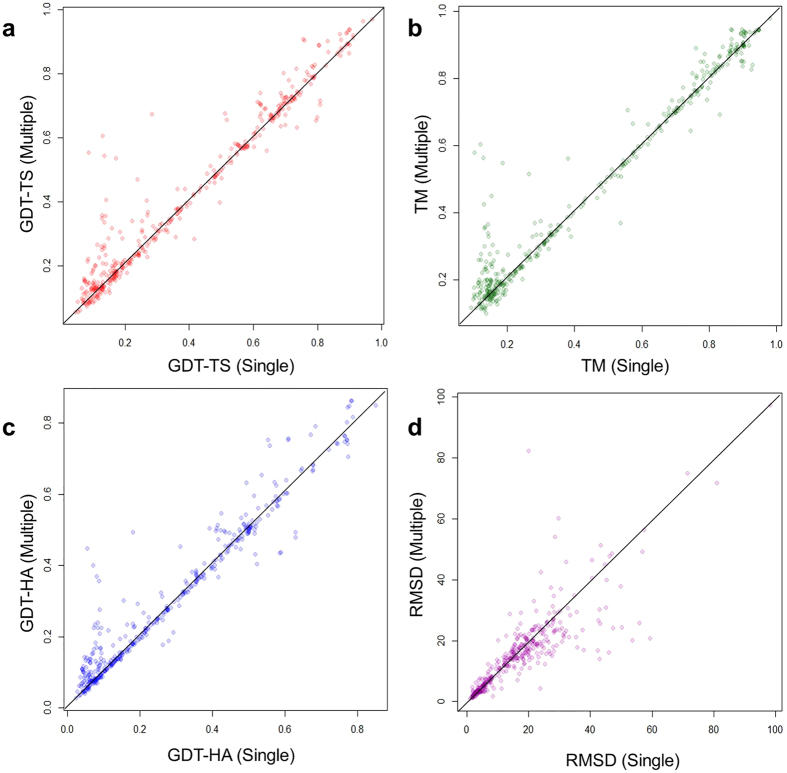
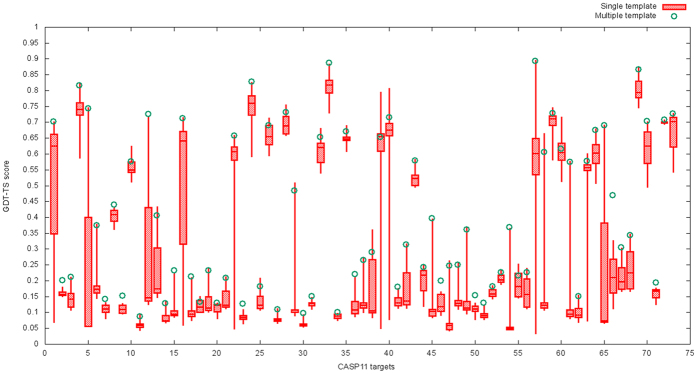
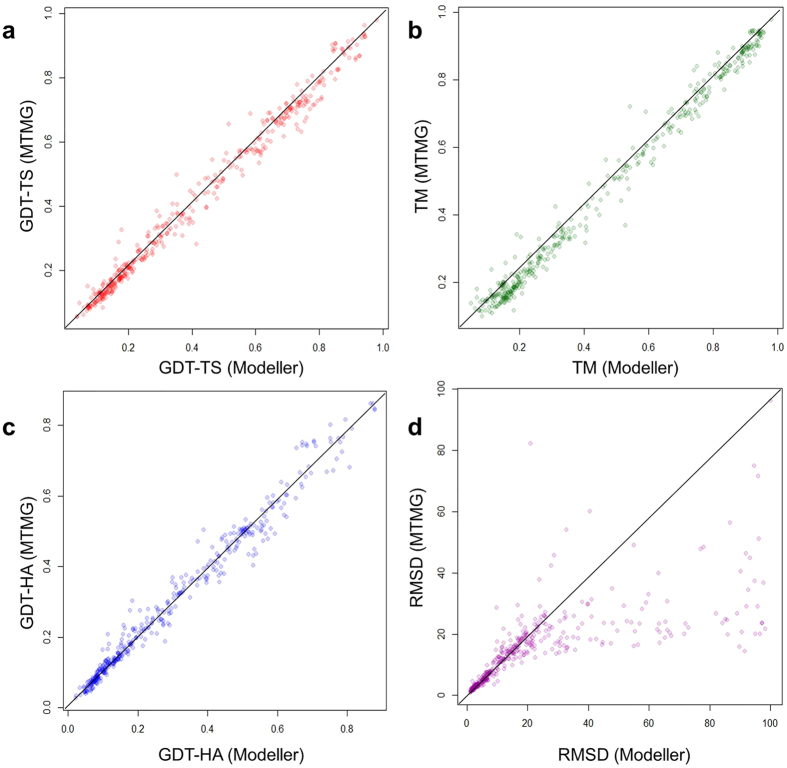
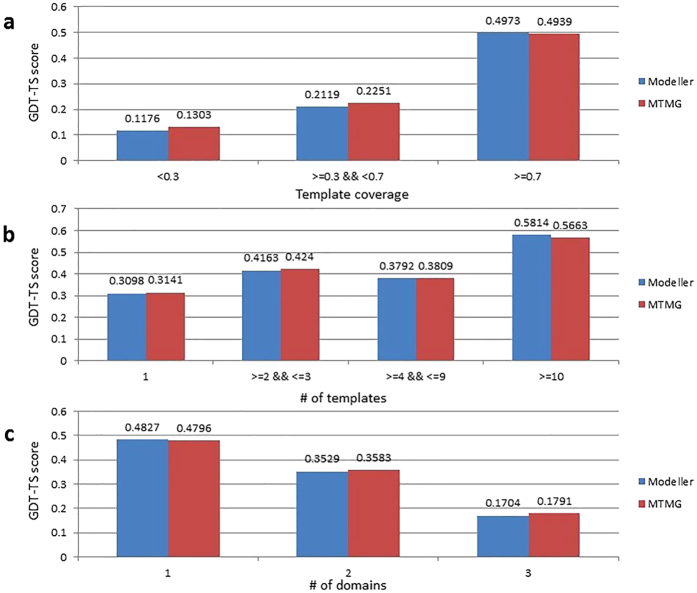
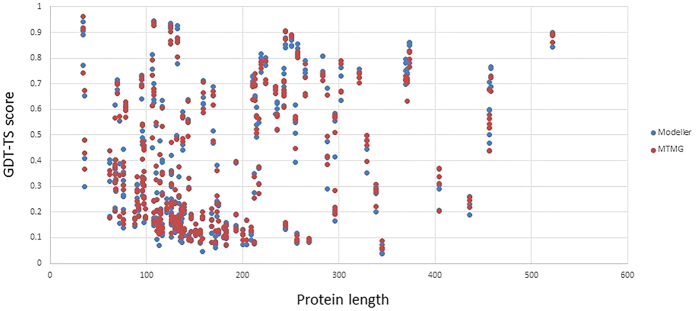
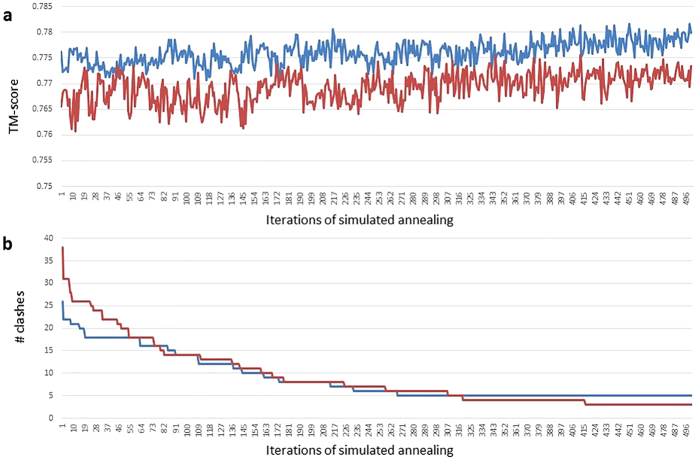
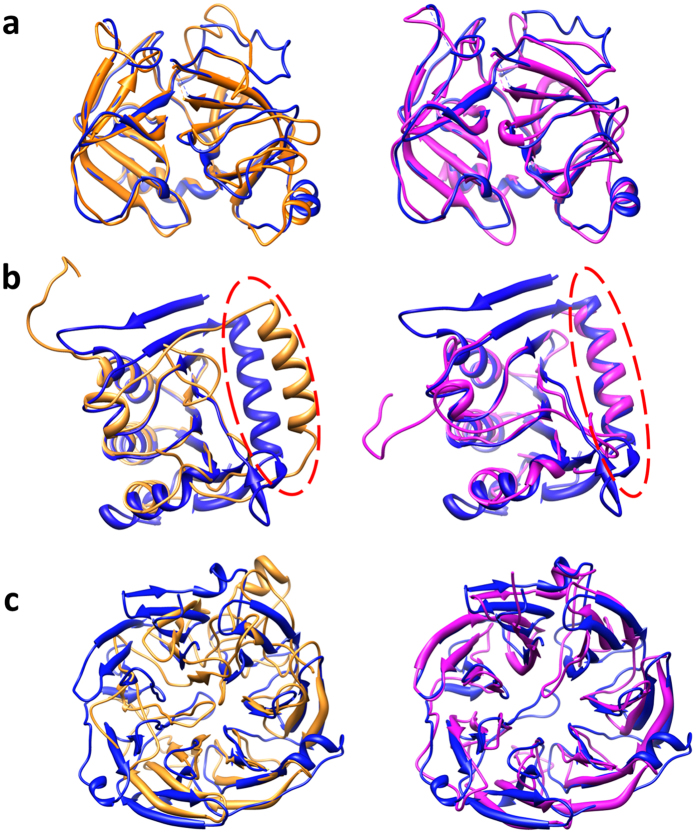
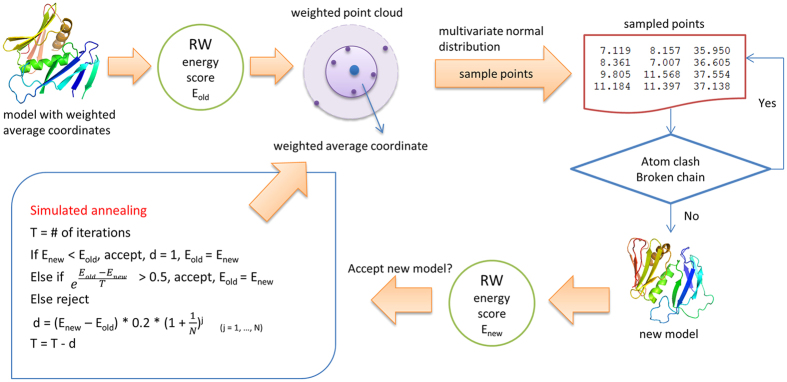
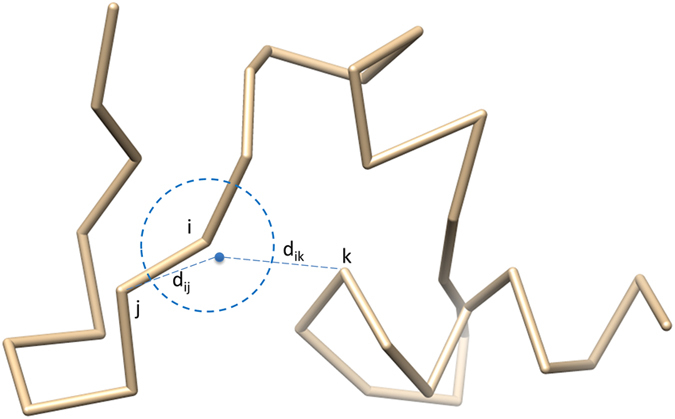
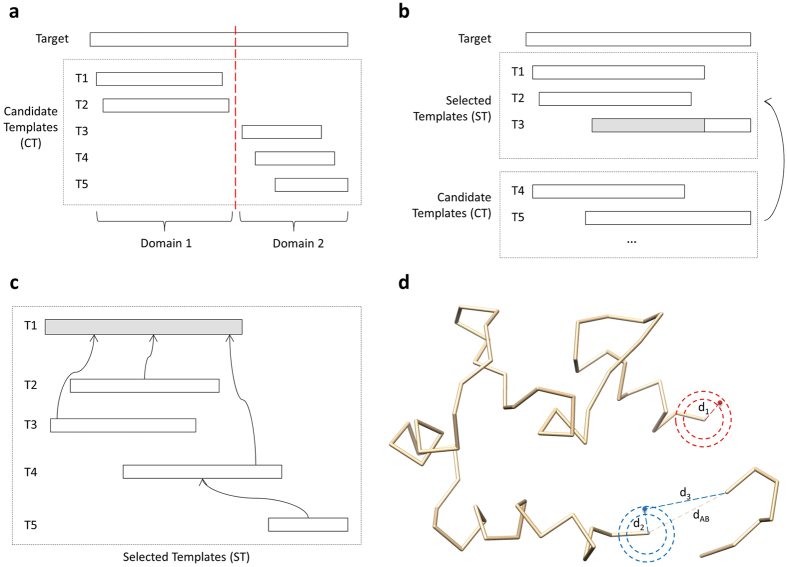
Generating tertiary structural models for a target protein from the known structure of its homologous template proteins and their pairwise sequence alignment is a key step in protein comparative modeling. Here, we developed a new stochastic point cloud sampling method, called MTMG, for multi-template protein model generation. The method first superposes the backbones of template structures, and the Cα atoms of the superposed templates form a point cloud for each position of a target protein, which are represented by a three-dimensional multivariate normal distribution. MTMG stochastically resamples the positions for Cα atoms of the residues whose positions are uncertain from the distribution, and accepts or rejects new position according to a simulated annealing protocol, which effectively removes atomic clashes commonly encountered in multi-template comparative modeling. We benchmarked MTMG on 1,033 sequence alignments generated for CASP9, CASP10 and CASP11 targets, respectively. Using multiple templates with MTMG improves the GDT-TS score and TM-score of structural models by 2.96-6.37% and 2.42-5.19% on the three datasets over using single templates. MTMG's performance was comparable to Modeller in terms of GDT-TS score, TM-score, and GDT-HA score, while the average RMSD was improved by a new sampling approach. The MTMG software is freely available at: http://sysbio.rnet.missouri.edu/multicom_toolbox/mtmg.html.
Figures
Similar articles
-
Assessment of template-based modeling of protein structure in CASP11.Proteins. 2016 Sep;84 Suppl 1(Suppl 1):200-20. doi: 10.1002/prot.25049. Epub 2016 Jun 15. Proteins. 2016. PMID: 27081927 Free PMC article.
-
Refined template selection and combination algorithm significantly improves template-based modeling accuracy.J Bioinform Comput Biol. 2019 Apr;17(2):1950006. doi: 10.1142/S0219720019500069. J Bioinform Comput Biol. 2019. PMID: 31057073
-
Template based protein structure modeling by global optimization in CASP11.Proteins. 2016 Sep;84 Suppl 1:221-32. doi: 10.1002/prot.24917. Epub 2015 Sep 14. Proteins. 2016. PMID: 26329522
-
Contact-assisted protein structure modeling by global optimization in CASP11.Proteins. 2016 Sep;84 Suppl 1:189-99. doi: 10.1002/prot.24975. Epub 2016 Jan 11. Proteins. 2016. PMID: 26677100
-
Template-based protein structure modeling.Methods Mol Biol. 2010;673:73-94. doi: 10.1007/978-1-60761-842-3_6. Methods Mol Biol. 2010. PMID: 20835794 Free PMC article. Review.
Cited by
-
DeepQA: improving the estimation of single protein model quality with deep belief networks.BMC Bioinformatics. 2016 Dec 5;17(1):495. doi: 10.1186/s12859-016-1405-y. BMC Bioinformatics. 2016. PMID: 27919220 Free PMC article.
-
Improving protein tertiary structure prediction by deep learning and distance prediction in CASP14.Proteins. 2022 Jan;90(1):58-72. doi: 10.1002/prot.26186. Epub 2021 Jul 27. Proteins. 2022. PMID: 34291486 Free PMC article.
References
-
- Eisenhaber F., Persson B. & Argos P. Protein structure prediction: recognition of primary, secondary, and tertiary structural features from amino acid sequence. Crit. Rev. Biochem. Mol. Biol. 30, 1–94 (1995). - PubMed
-
- Rost B. Protein structure prediction in 1D, 2D, and 3D. The Encyclopaedia of Computational Chemistry 3, 2242–2255 (1998).
-
- Floudas C. Computational methods in protein structure prediction. Biotechnol. Bioeng. 97, 207–213 (2007). - PubMed
-
- Wallner B., Fang H. & Elofsson A. Automatic consensus-based fold recognition using Pcons, ProQ, and Pmodeller. Proteins: Struct. Funct. Bioinform. 53, 534–541 (2003). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
