

The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes
- PMID: 27161491
- PMCID: PMC4866047
- DOI: 10.1038/ncomms11479
The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes
Abstract
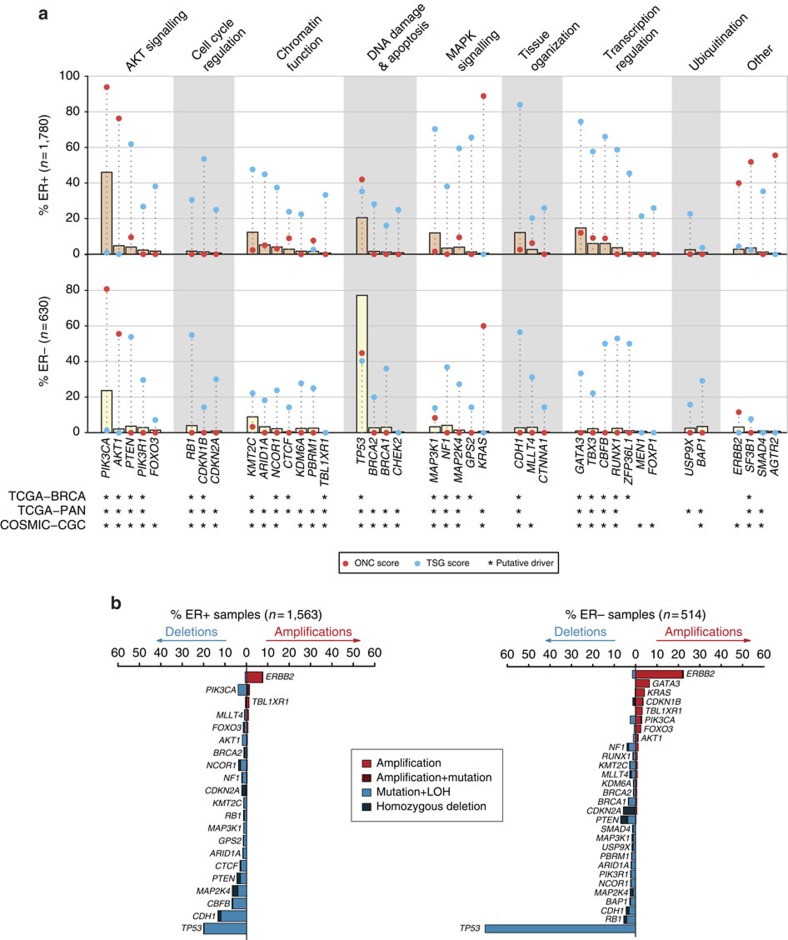
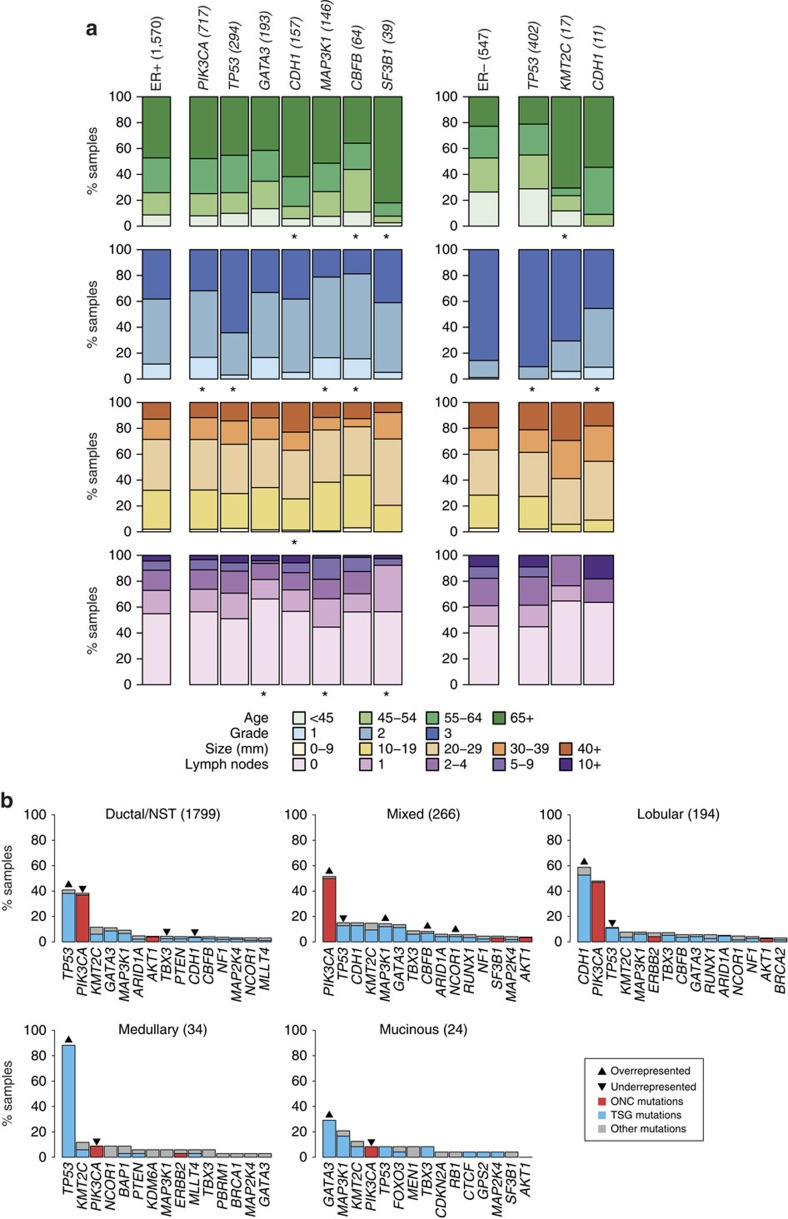
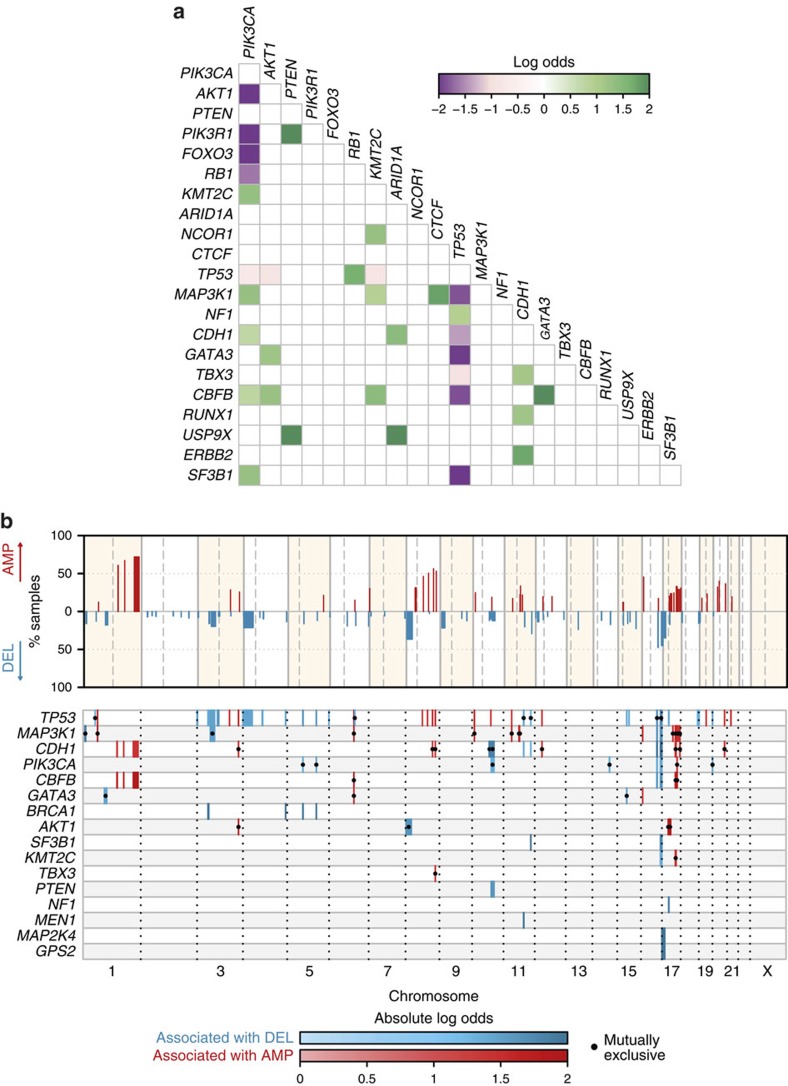
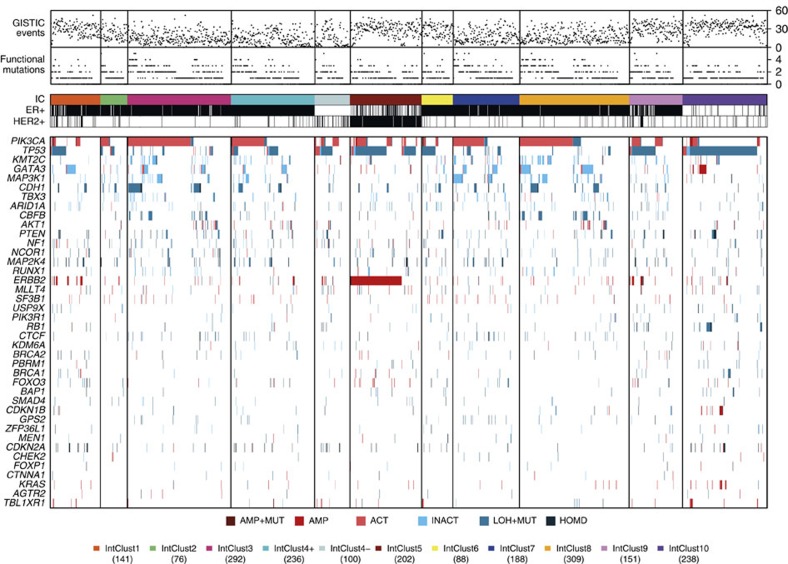
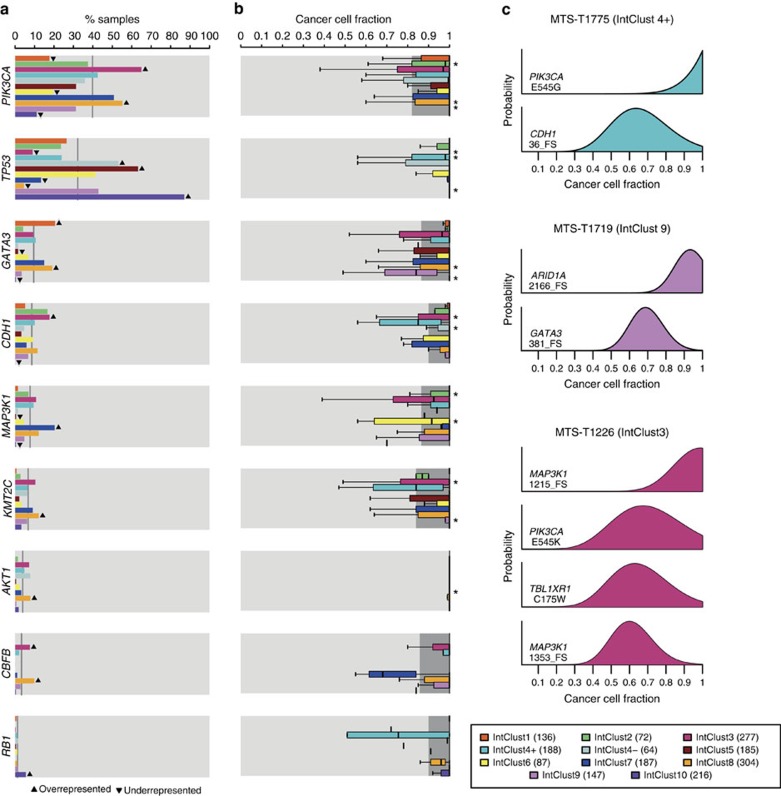
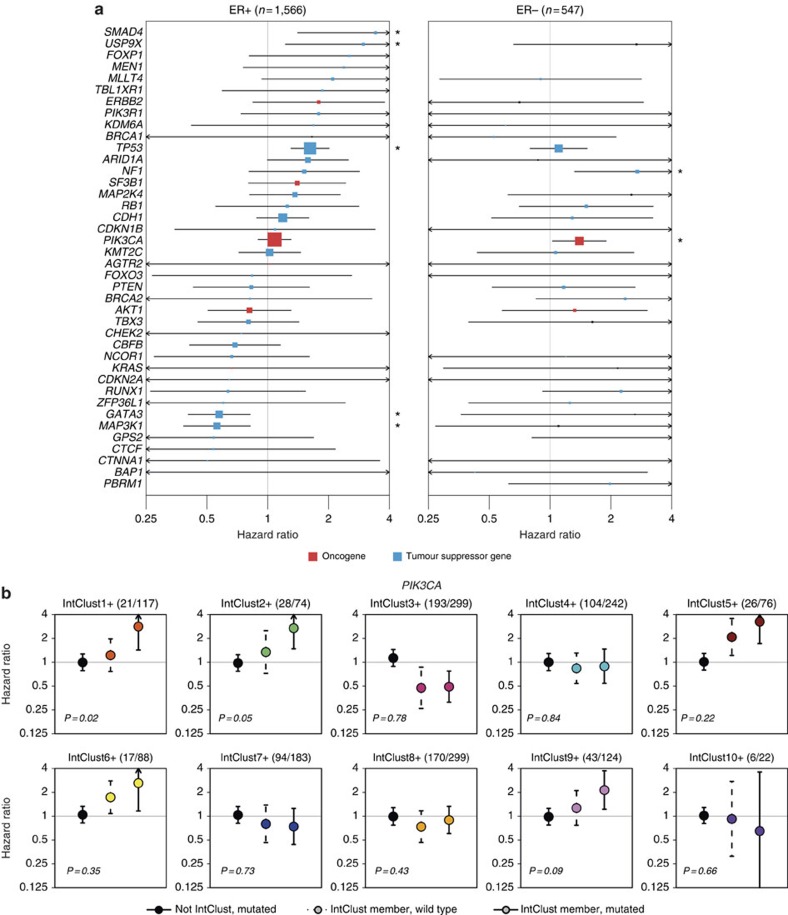
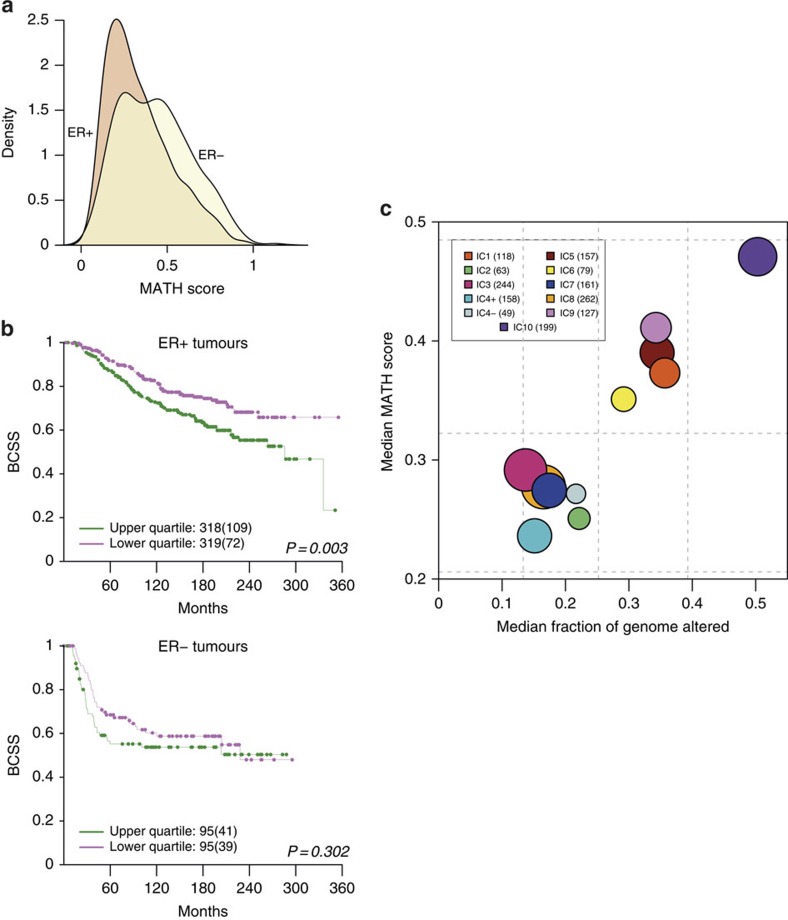
The genomic landscape of breast cancer is complex, and inter- and intra-tumour heterogeneity are important challenges in treating the disease. In this study, we sequence 173 genes in 2,433 primary breast tumours that have copy number aberration (CNA), gene expression and long-term clinical follow-up data. We identify 40 mutation-driver (Mut-driver) genes, and determine associations between mutations, driver CNA profiles, clinical-pathological parameters and survival. We assess the clonal states of Mut-driver mutations, and estimate levels of intra-tumour heterogeneity using mutant-allele fractions. Associations between PIK3CA mutations and reduced survival are identified in three subgroups of ER-positive cancer (defined by amplification of 17q23, 11q13-14 or 8q24). High levels of intra-tumour heterogeneity are in general associated with a worse outcome, but highly aggressive tumours with 11q13-14 amplification have low levels of intra-tumour heterogeneity. These results emphasize the importance of genome-based stratification of breast cancer, and have important implications for designing therapeutic strategies.
Conflict of interest statement
Helen Northen, John F. Peden, David R. Bentley and Mark T. Ross are full-time employees of Illumina Inc. Nitzan Rosenfeld is the Co-Founder and Chief Scientific Officer of Inivata Ltd. Dana W.Y. Tsui has acted as a consultant for Inivata Ltd prior to her current affiliation. Michelle Pugh is an employee of Inivata Ltd. The remaining authors declare no financial interests.
Figures
References
-
- Aparicio S. & Caldas C. The implications of clonal genome evolution for cancer medicine. N. Engl. J. Med. 368, 842–851 (2013). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
