

Ubiquitin E3 ligase FIEL1 regulates fibrotic lung injury through SUMO-E3 ligase PIAS4
- PMID: 27162139
- PMCID: PMC4886359
- DOI: 10.1084/jem.20151229
Ubiquitin E3 ligase FIEL1 regulates fibrotic lung injury through SUMO-E3 ligase PIAS4
Abstract
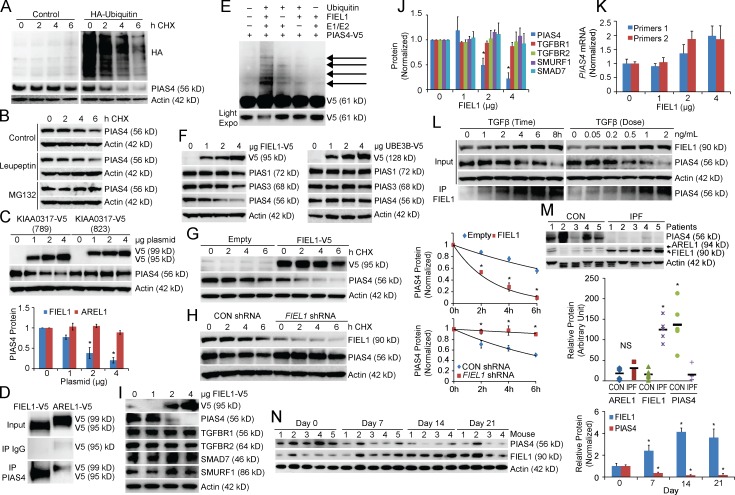
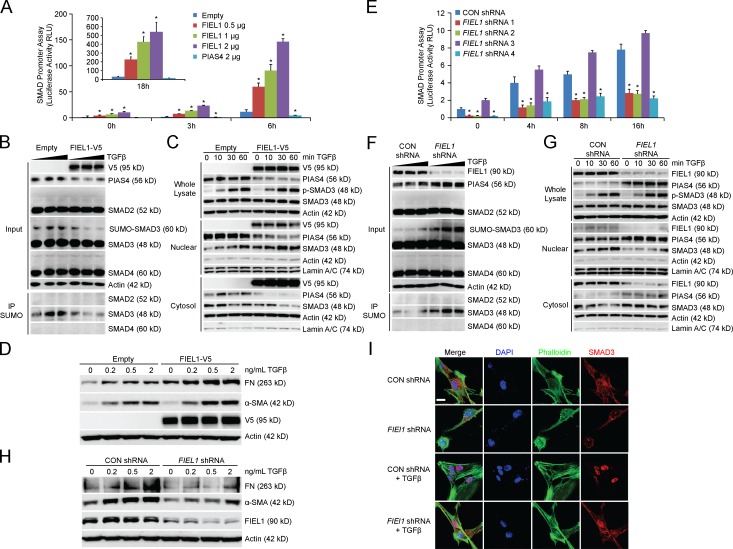
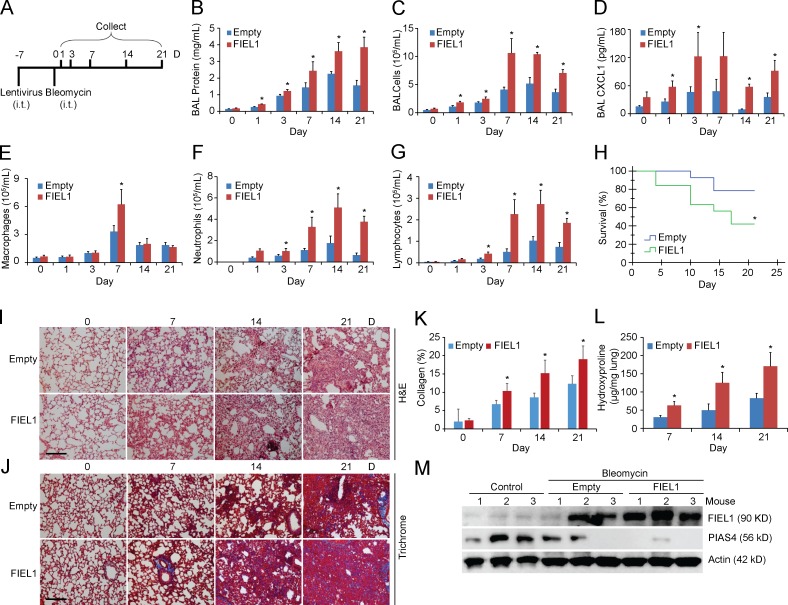
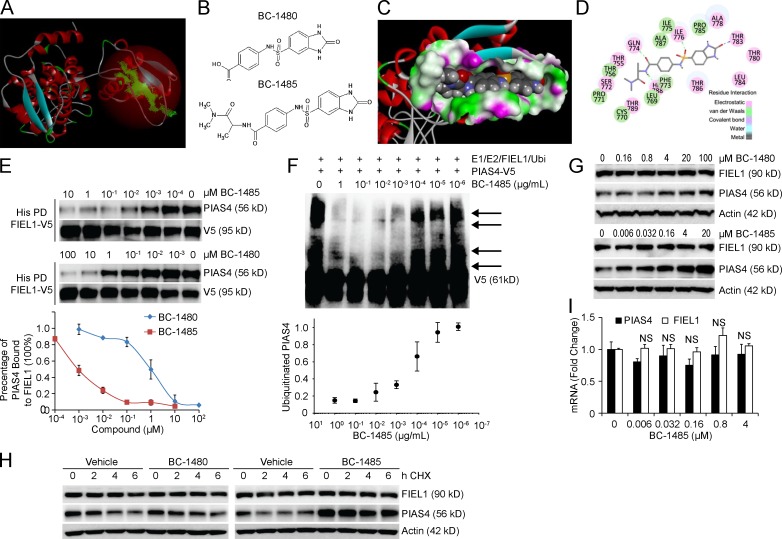
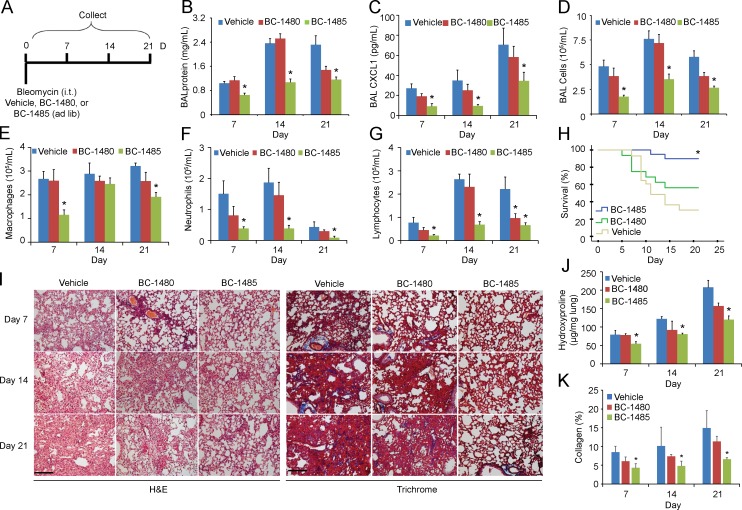
The E3 small ubiquitin-like modifier (SUMO) protein ligase protein inhibitor of activated STAT 4 (PIAS4) is a pivotal protein in regulating the TGFβ pathway. In this study, we discovered a new protein isoform encoded by KIAA0317, termed fibrosis-inducing E3 ligase 1 (FIEL1), which potently stimulates the TGFβ signaling pathway through the site-specific ubiquitination of PIAS4. FIEL1 targets PIAS4 using a double locking mechanism that is facilitated by the kinases PKCζ and GSK3β. Specifically, PKCζ phosphorylation of PIAS4 and GSK3β phosphorylation of FIEL1 are both essential for the degradation of PIAS4. FIEL1 protein is highly expressed in lung tissues from patients with idiopathic pulmonary fibrosis (IPF), whereas PIAS4 protein levels are significantly reduced. FIEL1 overexpression significantly increases fibrosis in a bleomycin murine model, whereas FIEL1 knockdown attenuates fibrotic conditions. Further, we developed a first-in-class small molecule inhibitor toward FIEL1 that is highly effective in ameliorating fibrosis in mice. This study provides a basis for IPF therapeutic intervention by modulating PIAS4 protein abundance.
© 2016 Lear et al.
Figures





References
-
- Akhmetshina A., Venalis P., Dees C., Busch N., Zwerina J., Schett G., Distler O., and Distler J.H.. 2009. Treatment with imatinib prevents fibrosis in different preclinical models of systemic sclerosis and induces regression of established fibrosis. Arthritis Rheum. 60:219–224. 10.1002/art.24186 - DOI - PubMed
-
- Baarsma H.A., Engelbertink L.H., van Hees L.J., Menzen M.H., Meurs H., Timens W., Postma D.S., Kerstjens H.A., and Gosens R.. 2013. Glycogen synthase kinase-3 (GSK-3) regulates TGF-β₁-induced differentiation of pulmonary fibroblasts. Br. J. Pharmacol. 169:590–603. 10.1111/bph.12098 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
