

Enzyme Sequestration as a Tuning Point in Controlling Response Dynamics of Signalling Networks
- PMID: 27163612
- PMCID: PMC4862689
- DOI: 10.1371/journal.pcbi.1004918
Enzyme Sequestration as a Tuning Point in Controlling Response Dynamics of Signalling Networks
Abstract
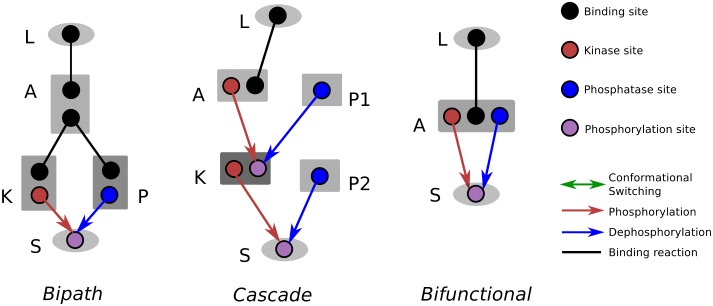
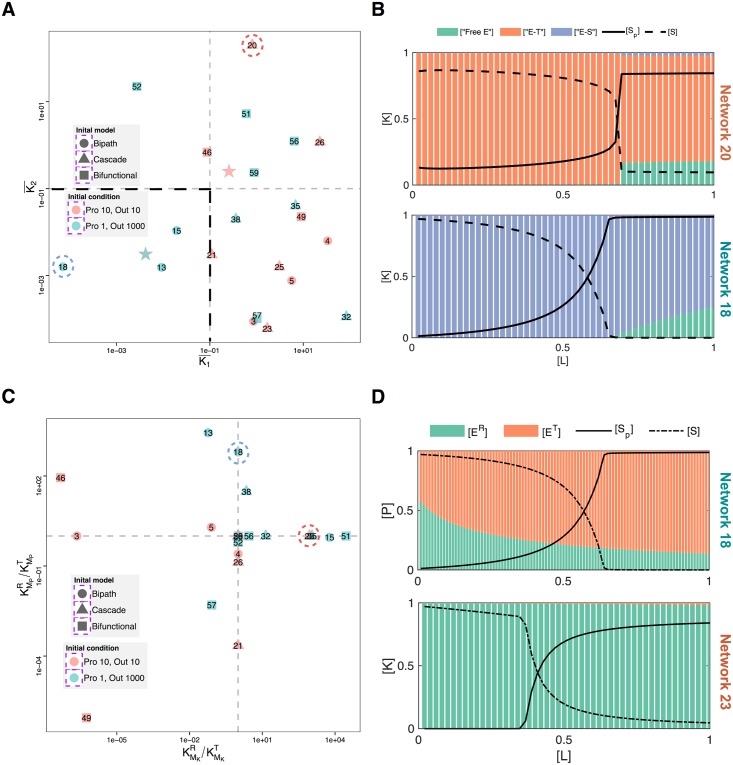
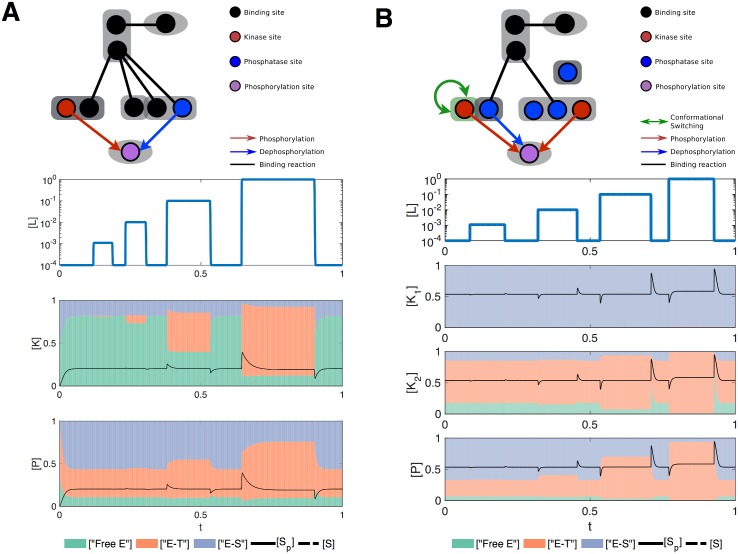
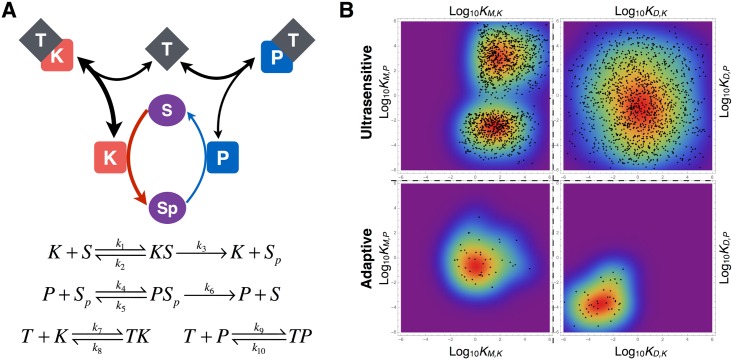
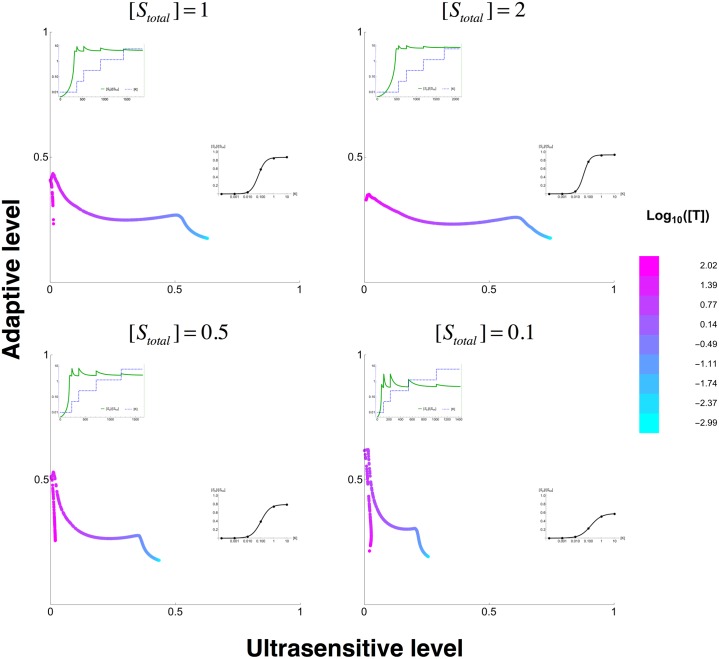
Signalling networks result from combinatorial interactions among many enzymes and scaffolding proteins. These complex systems generate response dynamics that are often essential for correct decision-making in cells. Uncovering biochemical design principles that underpin such response dynamics is a prerequisite to understand evolved signalling networks and to design synthetic ones. Here, we use in silico evolution to explore the possible biochemical design space for signalling networks displaying ultrasensitive and adaptive response dynamics. By running evolutionary simulations mimicking different biochemical scenarios, we find that enzyme sequestration emerges as a key mechanism for enabling such dynamics. Inspired by these findings, and to test the role of sequestration, we design a generic, minimalist model of a signalling cycle, featuring two enzymes and a single scaffolding protein. We show that this simple system is capable of displaying both ultrasensitive and adaptive response dynamics. Furthermore, we find that tuning the concentration or kinetics of the sequestering protein can shift system dynamics between these two response types. These empirical results suggest that enzyme sequestration through scaffolding proteins is exploited by evolution to generate diverse response dynamics in signalling networks and could provide an engineering point in synthetic biology applications.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Core signalling motif displaying multistability through multi-state enzymes.J R Soc Interface. 2016 Oct;13(123):20160524. doi: 10.1098/rsif.2016.0524. J R Soc Interface. 2016. PMID: 27733693 Free PMC article.
-
Computational models of signalling networks for non-linear control.Biosystems. 2013 May;112(2):122-30. doi: 10.1016/j.biosystems.2013.03.006. Epub 2013 Mar 14. Biosystems. 2013. PMID: 23499817
-
In Silico Evolution of Signaling Networks Using Rule-Based Models: Bistable Response Dynamics.Methods Mol Biol. 2019;1945:315-339. doi: 10.1007/978-1-4939-9102-0_15. Methods Mol Biol. 2019. PMID: 30945254
-
Model-based inference of biochemical parameters and dynamic properties of microbial signal transduction networks.Curr Opin Biotechnol. 2011 Feb;22(1):109-16. doi: 10.1016/j.copbio.2010.09.014. Epub 2010 Oct 20. Curr Opin Biotechnol. 2011. PMID: 20970318 Review.
-
Evolutionary principles underlying structure and response dynamics of cellular networks.Adv Exp Med Biol. 2012;751:225-47. doi: 10.1007/978-1-4614-3567-9_11. Adv Exp Med Biol. 2012. PMID: 22821461 Review.
Cited by
-
Enzyme sequestration by the substrate: An analysis in the deterministic and stochastic domains.PLoS Comput Biol. 2018 May 17;14(5):e1006107. doi: 10.1371/journal.pcbi.1006107. eCollection 2018 May. PLoS Comput Biol. 2018. PMID: 29771922 Free PMC article.
-
Exploring the mono-/bistability range of positively autoregulated signaling systems in the presence of competing transcription factor binding sites.PLoS Comput Biol. 2022 Nov 22;18(11):e1010738. doi: 10.1371/journal.pcbi.1010738. eCollection 2022 Nov. PLoS Comput Biol. 2022. PMID: 36413575 Free PMC article.
-
Core signalling motif displaying multistability through multi-state enzymes.J R Soc Interface. 2016 Oct;13(123):20160524. doi: 10.1098/rsif.2016.0524. J R Soc Interface. 2016. PMID: 27733693 Free PMC article.
-
Dynamics and Sensitivity of Signaling Pathways.Curr Pathobiol Rep. 2022 Jun;10(2):11-22. doi: 10.1007/s40139-022-00230-y. Epub 2022 Jun 27. Curr Pathobiol Rep. 2022. PMID: 36969954 Free PMC article.
-
NAIL: an evolutionarily conserved lncRNA essential for licensing coordinated activation of p38 and NFκB in colitis.Gut. 2021 Oct;70(10):1857-1871. doi: 10.1136/gutjnl-2020-322980. Epub 2020 Nov 25. Gut. 2021. PMID: 33239342 Free PMC article.
References
-
- Tyson JJ, Chen KC, Novak B. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr Opin Cell Biol. 2003;15: 221–231. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
